
Résumé
Les atteintes neurologiques de la maladie de Sjögren sont un défi en termes de diagnostic et de traitement (1). En effet, les manifestations sont variées et de sévérités très différentes.
Les atteintes périphériques sont les plus fréquentes avec les mononeuropathies crâniales, la neuropathie des petites fibres, les polyneuropathies axonales essentiellement sensitives, les syndromes dysautonomiques ou encore des formes sévères de neuronopathies sensitives ou de polyradiculonévrites. Les atteintes centrales sont très rares et majoritairement des scléroses en plaques like, des neuromyélites optiques ou des myélites transverses. De surcroît, les vascularites cryoglobulinémiques ont également fréquemment des expressions neurologiques, mais qui ne seront pas traitées dans ce document. Souvent l’atteinte neurologique précède de 3 à 5 ans les manifestations cliniques du Sjögren. Les mécanismes pathologiques, excepté pour la vascularite cryoglobulinémique, sont peu connus avec des hypothèses évoquant le rôle d’infiltrat lymphocytaire T ou de vascularite des vasa vasorum. Les principales manifestations systémiques sont décrites dans l’ESSDAI (Eular Sjögren Syndrome Disease Activity index) (2), qui permet d’estimer l’activité de la maladie. Le traitement des atteintes neurologiques reste difficile, car peu étudié et hétérogène. Le PNDS sur le Sjögren et la publication qui en a découlé décrivent les propositions thérapeutiques qui reposent surtout sur des recommandations d’experts (3-4).
Abstract
Neurological involvement in Sjögren’s disease: how to diagnose and treat it?
Neurological involvement in Sjögren disease presents a challenge in terms of diagnosis and treatment (1). Indeed, the manifestations are varied and of widely differing severity. Peripheral involvement is the most frequent, including cranial mononeuropathies, small fiber neuropathy, primarily sensory axonal polyneuropathies, dysautonomic syndromes, and severe forms of sensory neuronopathies or polyradiculoneuropathies. Central involvement is very rare and mainly presents as multiple sclerosis-like syndromes, neuromyelitis optica, or transverse myelitis. Furthermore, cryoglobulinemic vasculitis also frequently presents with neurological manifestations, but these will not be discussed in this document. Often, neurological involvement precedes the clinical manifestations of Sjögren by 3 to 5 years. The pathological mechanisms, except for cryoglobulinemic vasculitis, are poorly understood, with hypotheses suggesting the role of T-lymphocyte infiltrate or vasa vasorum vasculitis. The main systemic manifestations are described in the ESSDAI (Eular Sjögren Syndrome Disease Activity Index) (2), which allows for the estimation of disease activity. The treatment of neurological involvement remains difficult due to limited research and heterogeneity. The PNDS (National Diagnostic and Care Protocol) for Sjögren disease and the resulting publication describes therapeutic approaches, which are primarily based on expert recommendations (3-4).
Les atteintes du système nerveux périphérique
Le système nerveux périphérique est composé du système nerveux somatique et du système autonome (sympathique et parasympathique).
Les atteintes du système nerveux périphérique sont les plus connues et les plus fréquentes dans le Sjögren et atteignent environ 20 % des patients (Tab. 1). Les manifestations sont très variées avec les neuropathies des nerfs crâniens (névralgie du trijumeau, du nerf facial), les polyneuropathies (grosses et petites fibres et polyradiculoneuropathies) et les neuropathies du système autonome (syndrome dysautonomique). Les patients peuvent aussi développer des neuronopathies très sévères. L’examen complémentaire le plus souvent réalisé est l’électroneuromyogramme (ENMG) qui permet d’objectiver les atteintes des fibres. Ces atteintes parfois peu sévères cliniquement peuvent affecter fortement la qualité de vie des patients (5-6). L’un des domaines du score ESSDAI (Eular Sjögren Syndrome Disease Activity index) (2) décrit les plus fréquentes atteintes neurologiques périphériques (Tab. 2).
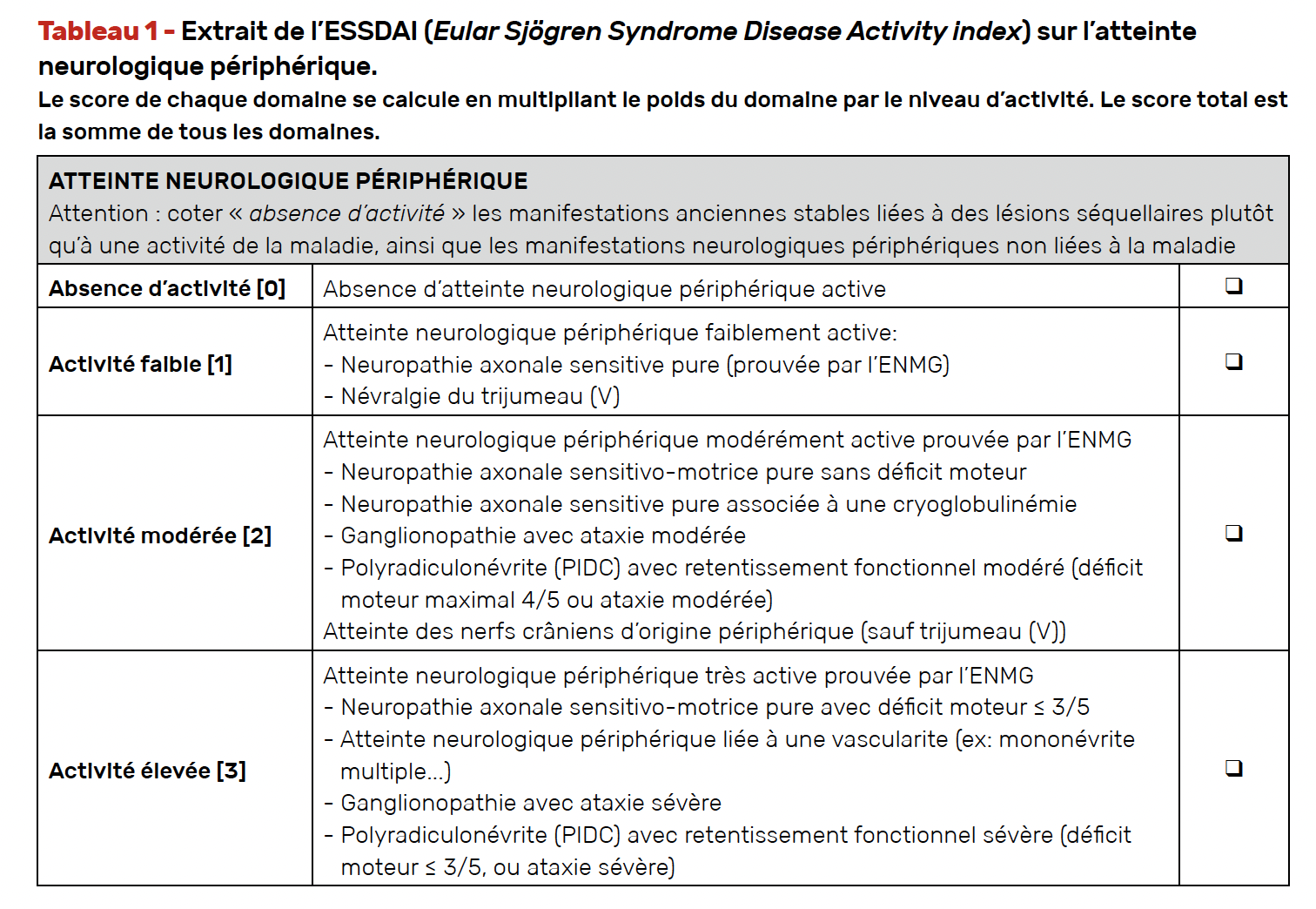
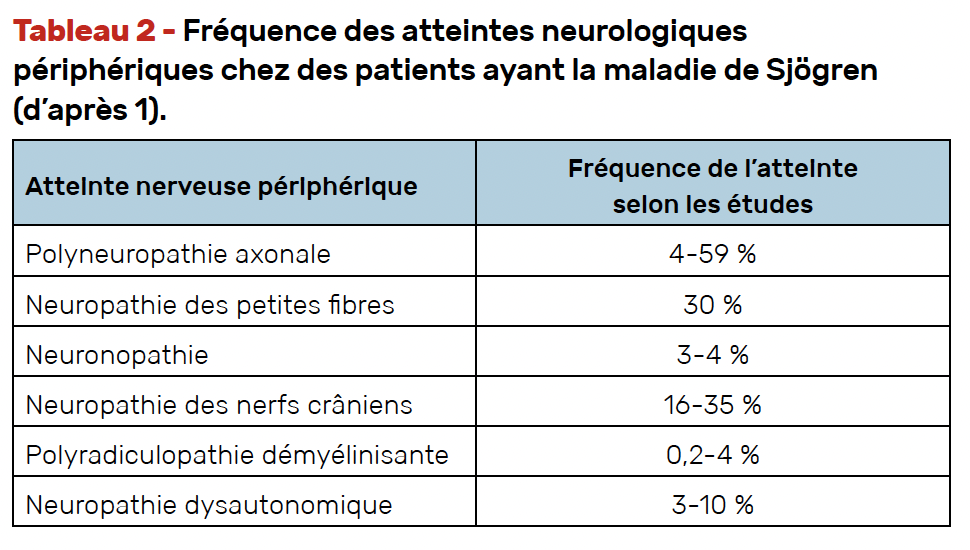
Le traitement des atteintes neurologiques reste difficile, car peu étudié et hétérogène. Le PNDS sur le Sjögren et la publication qui en a découlé, décrit les propositions thérapeutiques qui reposent surtout sur des recommandations d’experts (3-4).
Les mononeuropathies
Il s’agit de l’atteinte neurologique la plus connue de la maladie de Sjögren. L’atteinte des nerfs crâniens représenterait 20 % des atteintes périphériques, même si ces chiffres sont très variables en fonction des cohortes. Le nerf trijumeau est le plus fréquemment atteint, mais le nerf facial et le nerf vestibulo-cochléaire peuvent être aussi touchés.
Symptômes
Les symptômes sont en général une douleur unilatérale d’intensité variable à type de décharge électrique/coup de poignard/piqûre/élancement, dans le territoire du nerf atteint, de quelques secondes à 2 minutes, plusieurs fois par jour, provoquée par des stimuli normalement inoffensifs. Ces symptômes sont regroupés dans la classification IHCD3 permettant le diagnostic de névralgie du trijumeau (3-4).
Diagnostic
Un examen attentif permettra d’éliminer certains diagnostics différentiels (Lyme, sarcoïdose, herpès zona…). Une atteinte bilatérale doit faire évoquer une autre pathologie systémique.
Il est souvent recommandé de faire une IRM cérébrale afin d’éliminer un diagnostic différentiel (conflit vasculo-nerveux, une vascularite, une atteinte infectieuse ou tumorale).
Traitement et évolution
Le traitement est surtout fondé sur des antalgiques.
• En cas de symptômes invalidants, une corticothérapie à posologie moyenne, 0,2 à 0,3 mg/kg/j, peut être proposée pendant 10 jours (7).
• En cas d’atteinte faciale, un traitement par aciclovir peut se discuter.
L’évolution est souvent favorable sans séquelles.
Autres atteintes
D’autres atteintes sont possibles, mais plus rares, comme la mono-neuropathie des nerfs ulnaires ou médians, nécessitant également de réaliser un ENMG et d’éliminer d’autres causes compressives.
Les polyneuropathies axonales
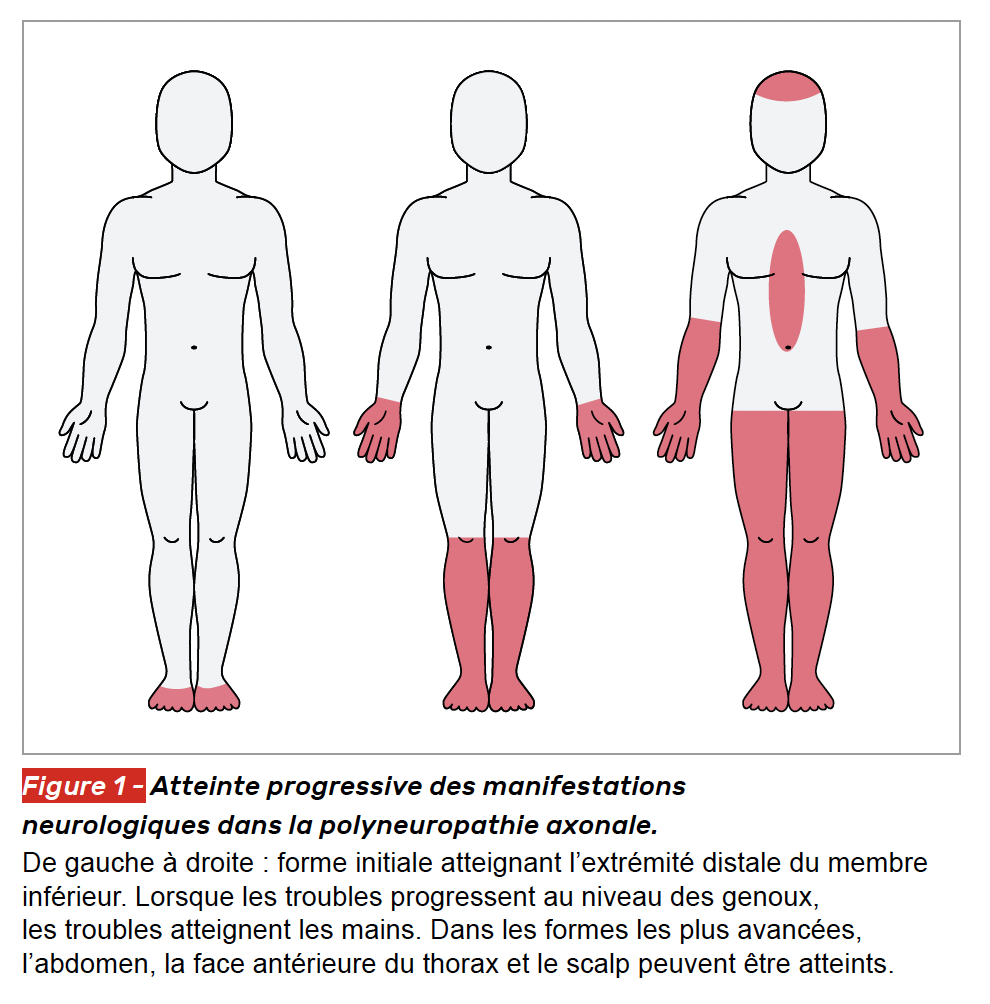
Une polyneuropathie se caractérise par une atteinte symétrique longueur dépendante et distale. Elle débute au niveau des pieds, puis, lorsqu’elle atteint les genoux, touche également les mains. Lorsqu’elle atteint les coudes, les fibres de l’abdomen et du scalp sont touchées (Fig. 1). Elles font partie des atteintes neurologiques fréquentes de la maladie de Sjögren, mais essentiellement sensitives (20 % des atteintes neurologiques) (5-6).
Symptômes
Dans le Sjögren, il s’agit surtout de formes sensitives pures associant paresthésies, dysesthésies, et des brûlures débutant au niveau des pieds.
L’atteinte peut être plus rarement sensitivo-motrice, avec abolition des réflexes, manque de force, ou steppage par déficit symétrique des releveurs du pied.
Diagnostic
Il est primordial dans ces cas de polyneuropathie de faire un bilan exhaustif des diagnostics différentiels tels qu’un diabète, une amylose, une iatrogénie ou encore une intoxication alcoolique chronique et d’avoir un avis neurologique.
L’ENMG peut être normal, mais peut aussi retrouver une diminution des potentiels sensitifs et des signes de dénervation.
L’origine axonale du trouble orienterait vers une atteinte ischémique. Une biopsie du nerf peut, dans certains cas, mettre en évidence une vascularite des vasa vasorum, par infiltrats ou encore par nécrose fibrinoïde.
Traitement
Le traitement reste symptomatique par des inhibiteurs de recapture de la sérotonine (fluoxétine), des antidépresseurs tricycliques, ou par gabapentine dans les formes peu sévères.
Si la forme est plus sévère, et notamment avec une atteinte motrice, un traitement par corticoïdes est toujours proposé en associant éventuellement à un autre immunosuppresseur et, en premier lieu, les IgIV. Cette prise en charge est à discuter au cas par cas au sein des centres de référence/compétences lors de réunion de concertation pluridisciplinaire (RCP). Un essai vient de se terminer en France, évaluant l’effet des IgIV dans ce contexte (essai TINISS – Pr Gottenberg).
La neuropathie des petites fibres
La neuropathie des petites fibres (NPF) est une atteinte des petites fibres myélinisées A-delta et des fibres amyéliniques C, responsables de la sensibilité thermo-algique, alors que les plus grosses fibres sensitives A-bêta ne sont pas affectées. La NPF est fréquemment décrite dans le Sjögren, où elle affecterait jusqu’à 30 % des patients (7-8). C’est une entité qui a été très largement décrite au cours des 20 dernières années et dont les causes potentielles sont multiples, en dehors du Sjögren, avec des maladies génétiques (amylose à transthyrétine), maladies métaboliques (diabète, carence en vitamine B12…), inflammatoires (gammapathie) et, dans 50 %, des formes finalement idiopathiques.
Symptômes
Elle atteint plutôt les Sjögren ayant des formes non systémiques et s’associe parfois à des symptômes de syndrome dysautonomique. Les patients ressentent surtout des sensations de brûlure, de douleur ou, au contraire, d’hypoesthésie ou d’hypoalgie, assez mal systématisées, couvrant souvent deux territoires distincts.
Diagnostic
Plusieurs scores d’évaluation peuvent être utilisés, le SFN-SIQ (small fiber neuropathy-symptom inventory questionnaire, utilisable pour le diagnostic), le DN4 (questionnaire douleur neuropathique en quatre questions), ou le COMPASS (composite autonomic symptom score, évaluant le syndrome dysautonomique).
L’ENMG est toujours normal lors d’une neuropathie des petites fibres.
Le diagnostic est complexe, afin de le confirmer, il existe plusieurs méthodes :
• mesure des seuils sensitifs thermiques,
• évaluation des potentiels évoqués nociceptifs généralement produits par stimulation laser
• ou encore une biopsie cutanée. Cette dernière permet l’analyse de la densité et de la quantité des fibres nerveuses intra-épidermiques, par marquage en immunofluorescence de la protéine PGP9.5. La quantité de fibres nerveuses intra-dermiques basse confirmerait le diagnostic.
Traitement
Actuellement, le traitement est uniquement symptomatique par antidépresseurs ou antiépileptiques. Il est important d’éviter toute escalade thérapeutique. Des traitements locaux par lidocaïne peuvent être proposés.
Les neuropathies dysautonomiques
Cette atteinte du système nerveux autonome est classiquement décrite dans le Sjögren, mais sa fréquence est inconnue. Rare, lorsqu’on la diagnostique sur des arguments objectifs, mais assez fréquemment rapportée par les patients à l’interrogatoire.
Symptômes
Les manifestations sont polymorphes : hypotension orthostatique, diarrhées, sudations, trouble du rythme cardiaque, troubles vésicaux…
Il existe une association avec la neuropathie des petites fibres ou avec la neuropathie sensitive.
Diagnostic
Le principal diagnostic étiologique différentiel de la neuropathie dysautonomique est le diabète. Le diagnostic nécessite d’avoir un avis spécialisé selon la fonction atteinte.
La cause reste inconnue, on évoque le rôle d’anticorps anti-récepteurs muscariniques de type 3. En effet, les récepteurs muscariniques de type 3 sont des récepteurs cholinergiques présents sur les cellules des glandes exocrines (salivaires, lacrymales) et aussi sur certaines structures du système nerveux autonome. Ils permettent la sécrétion glandulaire, mais aussi l’activité parasympathique autonome. Les anticorps anti-récepteurs muscariniques de type 3 bloquent ces récepteurs, et donc la sécrétion glandulaire et la transmission autonome.
Traitement
Le traitement est en général uniquement symptomatique et à discuter avec chacun des spécialistes d’organe (3-4).
La neuronopathie sensitive
La neuronopathie (anciennement appelée ganglionopathie sensitive) est une forme rare (environ 5 % des patients) et potentiellement très grave dans le Sjögren, résistante actuellement aux thérapeutiques (9). Elle précède, dans la majorité des cas, le début du Sjögren. Elle résulte d’une destruction des neurones sensitifs dans le ganglion rachidien postérieur responsable d’un handicap sévère du fait de l’ataxie et des douleurs qui en résultent.
Symptômes
C’est une neuropathie majoritairement non longueur dépendante (10) touchant les quatre membres, parfois la face, le thorax ou l’abdomen. Lorsque l’étiologie est auto-immune, comme dans la maladie de Sjögren, l’atteinte est généralement multifocale asymétrique, marquée par un infiltrat lymphocytaire T CD8.
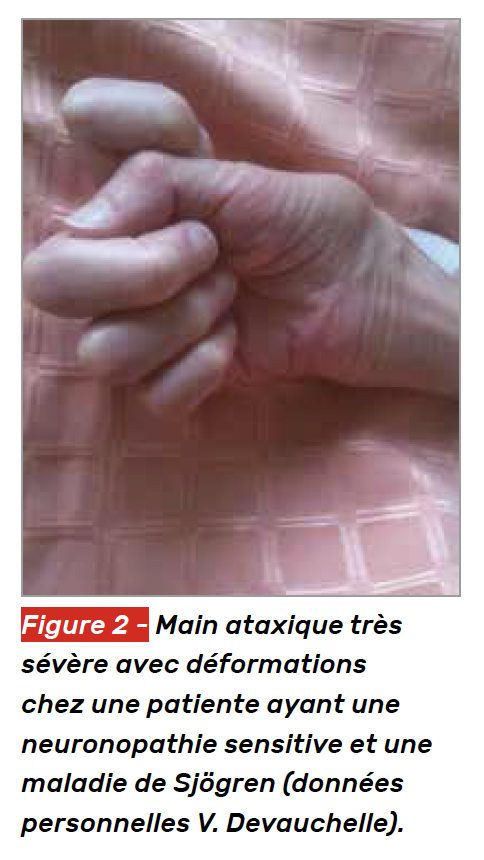
La présentation clinique typique débute par l’ataxie et des déficits sensitifs multifocaux et asymétriques qui différencient la neuronopathie des autres neuropathies motrices. Quand l’atteinte proprioceptive est majeure, les patients développent une pseudo-athétose des doigts et des orteils (empêchant la station debout ou l’utilisation de leurs mains) (Fig. 2). Les symptômes peuvent également s’accompagner de douleurs neuropathiques.
Le syndrome CANVAS
Lorsque l’ataxie est sévère, il est important de chercher un syndrome CANVAS (Cerebellar ataxia, neuropathy, and vestibular areflexia syndrome), caractérisé par une ataxie cérébelleuse (avec troubles de la coordination, l’équilibre et la parole), une neuropathie et une aréflexie vestibulaire. La mutation responsable de ce syndrome est une anomalie bi-allélique avec expansions dans l’intron de RFC1. Ce syndrome est extrêmement rare, mais il survient souvent après 50 ans et est d’évolution lentement progressive. Il faut cependant y penser devant l’association de ces trois atteintes (cérébelleuse, neuropathie et syndrome vestibulaire) (11).
Diagnostic
Lors du bilan de la neuronopathie, la mise en évidence des anticorps anti-Hu permet d’évoquer une origine paranéoplasique (qui a été à l’origine des premières descriptions en 1948 des neuronopathies satellites de cancers pulmonaires), tandis que les anticorps anti-FGFR3 et anti-AGO traduiraient une origine dysimmunitaire, mais ils ne sont pas disponibles en routine. Les autres causes principales sont toxiques (vitamine B, chimiothérapie) ou infectieuses.
À l’ENMG, une atteinte sévère et diffuse des potentiels d’action sensitifs avec un respect de la conduction motrice est très évocatrice de neuronopathie sensitive.
L’IRM des ganglions postérieurs cherche à visualiser leur atteinte dégénérative, mais reste souvent non informative.
Comme aide au diagnostic, il existe le score de Camdessanché. Le diagnostic de neuronopathie sensitive est possible si le score est > 6,5. Le diagnostic est notamment considéré comme probable si le score est > 6,5 et qu’une maladie de Sjögren est associée.
Traitement
Le traitement est difficile. Dans les formes sévères, il est possible d’avoir recours à une corticothérapie par bolus, à des IgIV (2 g/kg), échanges plasmatiques, immunosuppresseurs par cyclophosphamide ou par mycophénolate mofétil.
Il est recommandé d’avoir rapidement recours aux centres experts et de proposer des traitements agressifs d’emblée, même en l’absence de preuves scientifiques.
Les polyradiculonévrites inflammatoires démyélinisantes chroniques
La polyradiculonévrite inflammatoire démyélinisante chronique est sensitivo-motrice. Elle touche les racines nerveuses et les nerfs les prolongeant, entraînant une faiblesse musculaire avec sensations altérées, des réflexes ostéo-tendineux réduits, voire abolis.
Il s’agit d’une atteinte rare, mais très sévère du Sjögren.
Traitement
Les recommandations thérapeutiques actuelles préconisent un traitement par corticoïdes et par Ig IV 2 g/kg sur 4 jours, toutes les 4 semaines en induction et extension.
Les atteintes du système nerveux central
Ce sont des atteintes très rares (1-5 %), mais, bien sûr, beaucoup plus sévères que les atteintes périphériques, et souvent plus invalidantes (Tab. 3).
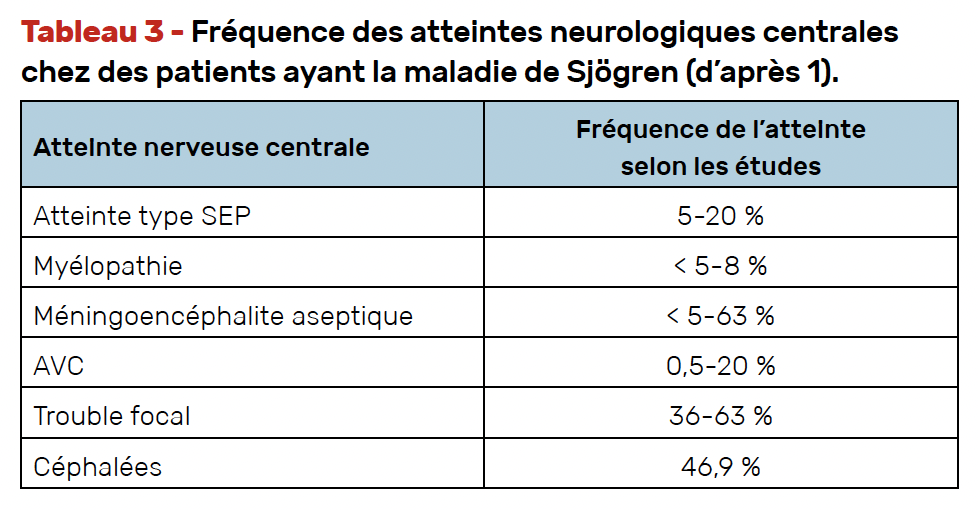
Il n’y a quasiment aucune étude les concernant spécifiquement dans le Sjögren et leur traitement, en dehors de la vascularite cryoglobulinémique, est fondé sur des discussions entre neurologues et rhumatologues. L’un des domaines du score ESSDAI (2) décrit les plus fréquentes atteintes neurologiques centrales (Tab. 4).

L’atteinte de type sclérose en plaques (12)
Les patients peuvent alors présenter des symptômes de sclérose en plaques (SEP) : parésies, aphasie, ataxie, ophtalmoplégie internucléaire…
Diagnostic
Il est possible de retrouver des lésions de la substance blanche au niveau périventriculaire, juxta-cortical, sous-tentoriel ou médullaire à l’IRM (les lésions des corps calleux ou des noyaux gris centraux sont inhabituelles dans la SEP et pourraient s’intégrer dans ce concept de SEP like), ainsi que des bandes oligoclonales et un index IgG augmenté à la ponction lombaire.
Traitement
Les traitements sont les mêmes que dans la SEP, et le patient doit être géré par/avec les neurologues.
La neuromyélite optique ou son spectre (NMOSD/neuromyelitis optica spectrum disorders)
Maladie auto-immune démyélinisante, elle est associée dans 30 % des cas à une maladie auto-immune, dont la maladie de Sjögren.
Symptômes
Lors d’une neuromyélite optique, il est possible d’observer une névrite optique se manifestant par une douleur oculaire et une baisse de l’acuité visuelle rapide mono ou binoculaire. Une myélite peut entraîner une paralysie plus ou moins complète des membres, une hyper-algie, une anesthésie, ou encore une perte de contrôle des sphincters. Il peut également y avoir une atteinte du tronc cérébral se manifestant, par exemple, par des vomissements ou un hoquet.
Diagnostic
Il est important de doser systématiquement les anticorps anti-aquaporine 4 (anti-AQP4) lors d’une myélite optique, d’une myélite ou encore d’une maladie du spectre de la neuromyélite optique (NMOSD). Ils permettent de confirmer le diagnostic lorsque leur positivité est associée à une atteinte du système nerveux central. Si les anticorps sont négatifs, le diagnostic repose alors sur la manifestation de deux poussées et sur des anomalies à l’IRM.
Traitement et évolution
• Le traitement d’une poussée repose sur des corticoïdes en intraveineux.
• Le traitement de fond, primordial lors d’une positivité des anti-AQP4, est le même que pour une neuromyélite idiopathique : anti-CD20/CD19, anti-IL-6 récepteur ou anti-complément.
La myélite transverse/diffuse
Les patients présentent le plus souvent des paraparésies, tétraparésies, des troubles sensitifs et un dysfonctionnement du sphincter vésical.
Diagnostic et traitement
Une IRM permet d’éliminer un diagnostic différentiel, notamment un signal inflammatoire du ganglion postérieur vers la moelle dans un contexte de ganglionopathie. Un traitement par corticoïdes et cyclophosphamide est recommandé.
La méningo-encéphalite aseptique (12)
Bien qu’exceptionnelle, on peut citer la méningo-encéphalite aseptique comme possible atteinte de la maladie de Sjögren. Elle peut aussi être retrouvée dans la sarcoïdose, la maladie de Behçet, le lupus érythémateux systémique ou encore la granulomatose de Wegener. La méningo-encéphalite est dite “aseptique” lorsqu’aucune bactérie n’est retrouvée, ni à l’examen direct, ni à la culture du liquide céphalorachidien. Ce dernier serait à prédominance lymphocytaire, avec cellules plasmocytaires et une cellularité < 50/mm3.
Traitement
Une fois que les étiologies virales et bactériennes sont éliminées, les traitements probabilistes par aciclovir et antibiotiques peuvent être arrêtés. Le traitement repose sur des corticoïdes afin de diminuer l’inflammation.
Les dysfonctions cognitives
Rapportées dans de nombreuses maladies auto-immunes, les dysfonctions cognitives sont aussi assez souvent retrouvées dans le Sjögren. La physiopathologie est inconnue, peut-être liée à une endothélite cérébrale (mais non retrouvée sur des autopsies), à la présence d’auto-anticorps spécifiques (anti-NR2) ou à d’autres mécanismes non élucidés. La fréquence de lésions de la substance blanche semble plus fréquente chez les Sjögren, mais sans lien clinique évident et des analyses en SPECT cérébrale montrent un défaut de diffusion chez des Sjögren ayant des fonctions cognitives altérées.
Symptômes
Les symptômes sont très variés, mais les plus décrits sont des troubles de l’attention ou de la mémoire de travail. Ces troubles co-existent souvent avec la fatigue, la douleur et la dépression.

Les auteures déclarent ne pas avoir de liens d’intérêt en rapport avec cet article. Le contenu et/ou les opinions exprimées, notamment celui ou celle(s) relatifs à la stratégie thérapeutique, ont été réalisés en toute indépendance.
Bibliographie
1. Carvajal Alegria G, Guellec D, Devauchelle-Pensec V, Saraux A. Is there specific neurological disorders of primary Sjogren’s syndrome? Joint Bone Spine 2015 ; 82 : 86-9.
2. Seror R, Bowman SJ, Brito-Zeron P et al. EULAR Sjogren’s syndrome disease activity index (ESSDAI): a user guide. RMD Open 2015 ; 1 : e000022.
3. Devauchelle-Pensec V, Mariette X, Benyoussef AA et al. French national diagnostic and care protocol for Sjogren’s disease. Rev Med Interne 2023 ; 44 : 423-57.
4. HAS. Protocoles nationaux de diagnostic et de soins. Maladie (ou syndrome) de Sjögren. 2022.
5. Tobon GJ, Pers JO, Devauchelle-Pensec V, Youniou P. Neurological disorders in primary Sjogren’s syndrome. Autoimmune Dis 2012 ; 2012 : 645967.
6. Carvajal Alegria G, Guellec D, Mariette X et al. Epidemiology of neurological manifestations in Sjogren’s syndrome: data from the French ASSESS Cohort. RMD Open 2016 ; 2 : e000179.
7. Seeliger T, Neelke Dreyer H, Siemer JM et al. Clinical and paraclinical features of small fiber neuropathy in Sjögren’s syndrome. J Neurol 2023 ; 270 : 1004-10.
8. Nebuchennykh M, Løseth S, Lindal S et al. The value of skin biopsy with recording of intraepidermal nerve fiber density and quantitative sensory testing in the assessment of small fiber involvement in patients with different causes of polyneuropathy. J Neurol 2009 ; 256 : 1067-75.
9. Antoine JC. Les neuronopathies sensitives dysimmunes : enjeux diagnostiques et thérapeutiques. Bull Acad Nat Med 2021 ; 205 : 937-45.
10. Funakoshi T, Yamada M, Ikeda K et al. Sjögren’s syndrome with sensory neuronopathy and polyradiculoneuropathy. Intern Med 2025 ; 64 : 3033-8.
11. Cortese A, Simone R, Sullivan R et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet 2019 ; 51 : 649-58.
12. Camard M, Urbain F, Noel N. Cognitive impairment in systemic autoimmune and inflammatory diseases: A narrative review focused on ANCA-associated vasculitis, sarcoidosis, Sjögren’s syndrome, systemic sclerosis, and Behçet’s disease. Autoimmun Rev 2025 ; 24 : 103899.