Hémochromatose et arthropathie : les nouveaux critères de classification
Audrey Le Toux
Depuis le XIXe siècle, il a été observé une association entre surcharge en fer et atteintes articulaires. Cependant, le rôle du fer dans le métabolisme osseux, notamment en cas d’hémochromatose semble partiellement élucidé. On parle de surcharge en fer pour des taux de ferritine supérieurs à 200 µg/l chez la femme et 300 µg/l chez l’homme, avec un coefficient de saturation de la transferrine > 45 %.
Sources de fer et transport
Les deux principales sources de fer sont le recyclage des cellules sanguines par les macrophages et l’apport alimentaire. De nombreuses protéines, notamment la ferroportine, la céroplasmine ou la transferrine, sont impliquées dans son transport. Il est retrouvé des récepteurs du fer dans les cellules de lignées ostéoblastiques et ostéoclastiques expliquant l’implication du fer dans le métabolisme osseux (1, 2).
Toxicité du fer
L’effet délétère du fer est retrouvé pour des valeurs parfois en dessous du seuil de surcharge (> 5 μg in vitro (3)). Il est lié à une augmentation de la mortalité des ostéoblastes ainsi qu’à une diminution de leur production de collagène. Les ostéoclastes sont également impliqués avec une diminution de leur activité de minéralisation. La toxicité du fer est également mise en évidence dans la matrice osseuse avec une altération de la structure des cristaux d’hydroxyapatite (4). L’implication de la toxicité ferrique n’est toutefois pas équivalente selon les types de surcharge (génétique ou secondaire) et reste liée au fonctionnement de l’axe hepcidine-ferroportine (3).
• En effet, dans la surcharge génétique, il est retrouvé une accumulation tissulaire de fer avec une augmentation de l’activité de la ferroportine et une diminution de la concentration d’hepcidine.
• Dans les surcharges secondaires, il est retrouvé des modifications contraires avec une accumulation plutôt macrophagique de fer, une diminution de l’activité de la ferroportine ainsi qu’une augmentation de la concentration plasmatique d’hepcidine (3).
De plus, dans l’ostéoporose post-ménopausique en lien avec l’hypogonadisme induit par la surcharge, il semble exister une amélioration de la densité minérale osseuse (DMO) à tous les sites chez les patients traités par phlébotomie (5, 6).
Les atteintes articulaires
Les conséquences articulaires de la surcharge en fer sont principalement les douleurs et les déformations articulaires à cause d’érosions osseuses. Parmi les atteintes les plus typiques, nous retrouvons l’atteinte des métacarpophalangiennes (MCP) 2 et 3 avec limitation de la flexion, les ostéophytes “en hameçon” ou les kystes sous-chondraux. Ces atteintes ont parfois un phénotype très proche de celles du rhumatisme à cristaux de PPC ou de l’arthrose rendant le diagnostic parfois délicat. De plus, dans 25 % des cas, des signes de chondrocalcinose sont également retrouvés.
Les nouveaux critères de classification Eular
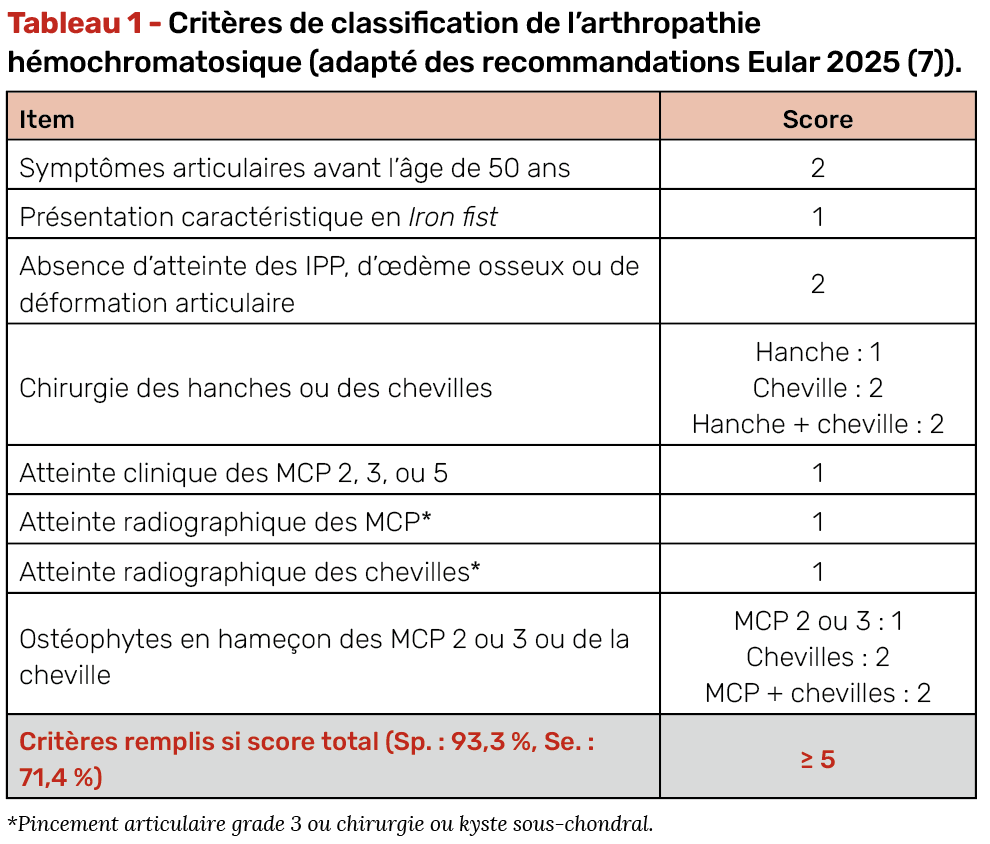
De récents critères de classification Eular ont été publiés en 2026 (7) et sont présentés dans le tableau 1. Ces critères sont applicables chez les patients pour guider le diagnostic d’arthropathie liée au métabolisme martial en cas :
1. de mutation homozygote C282Y du gène HFE,
2. de douleurs articulaires persistantes,
3. avec surcharge en fer (nécessité de réalisation d’au moins 16 phlébotomies ou coefficient de saturation de la transferrine > 45 % à deux reprises ou excès de fer visualisé sur biopsie hépatique).
Place de mutations mineures d’HFE
La question de la place des mutations “mineures” du gène HFE reste débattue bien qu’elles soient probablement impliquées dans l’apparition des arthropathies liées à la surcharge en fer.
Place de la phlébotomie
L’intérêt de la phlébotomie est mis en évidence dans les principaux organes touchés, mais ne permet pas de prévenir l’évolution sur le plan articulaire. En effet, la progression des atteintes est retrouvée même chez les patients déplétés en fer.
Place de l’imagerie
Différentes techniques d’imagerie peuvent être intéressantes pour l’évaluation de l’atteinte articulaire.
• La tomographie quantitative haute résolution (HR-pQCT) permet la mesure de densités volumétriques et l’évaluation de la microarchitecture osseuse, peut être utilisée pour visualiser les modifications du remodelage osseux, mais également pour l’étude plus précise du pincement articulaire (8).
• L’échographie peut mettre en évidence des synovites, des atteintes du cartilage et des dépôts de PPC (9).
• L’IRM retrouve exceptionnellement un œdème osseux (10). Il n’est pas visualisé de dépôt de fer à proprement parler dans les synoviales des patients atteints.
Ainsi, ces différentes techniques permettent de définir l’arthropathie hémochromatosique comme mixte avec une composante à la fois inflammatoire et dégénérative.
Finalement, il faut penser à une atteinte articulaire d’hémochromatose devant :
• des lésions sévères d’arthrose avant l’âge de 50 ans ;
• l’absence d’histoire traumatique ;
• l’atteinte des MCP et des chevilles ;
• les kystes florides, les ostéophytes et les pertes importantes de cartilage.
Bibliographie
1. Roodman GD. Osteoclasts pump iron. Cell Metab 2009 ; 9 : 405-6.
2. Kasai K, Hori MT, Goodman WG. Characterization of the transferrin receptor in UMR-106-01 osteoblast-like cells. Endocrinology 1990 ; 126 : 1742-9.
3. Robin F, Chappard D, Leroyer P et al. Differences in bone microarchitecture between genetic and secondary iron-overload mouse models suggest a role for hepcidin deficiency in iron-related osteoporosis. FASEB J 2023 ; 37 : e23245.
4. Guggenbuhl P, Filmon R, Mabilleau G et al. Iron inhibits hydroxyapatite crystal growth in vitro. Metabolism 2008 ; 57 : 903-10.
5. Hibbert EJ, Fulcher GR, Coyle L et al. Effect of venesection on bone mineral density in an eugonadal woman with haemochromatosis. J Gastroenterol Hepatol 1999 ; 14 : 17-8.
6. Diamond T, Stiel D, Posen S. Effects of testosterone and venesection on spinal and peripheral bone mineral in six hypogonadal men with hemochromatosis. J Bone Miner Res 1991 ; 6 : 39-43.
7. Kiely PD, Finzel S, Farisogullari B et al. EULAR 2025 classification criteria for haemochromatosis arthropathy. Ann Rheum Dis 2026 ; 85 : 238-45.
8. Heilmeier U, Burghardt AJ, Tse JJ et al. Analysis of hand joint space morphology in women and men with hereditary hemochromatosis. Calcif Tissue Int 2023 ; 112 : 440-51.
9. Dejaco C, Stadlmayr A, Duftner C et al. Ultrasound verified inflammation and structural damage in patients with hereditary haemochromatosis-related arthropathy. Arthritis Res Ther 2017 ; 19 : 243.
10. Frenzen K, Schäfer C, Keyßer, G. Erosive and inflammatory joint changes in hereditary hemochromatosis arthropathy detected by low-field magnetic resonance imaging. Rheumatol Int 2013 ; 33 : 2061-7.
Biomédicaments et risque de cancer dans les RIC : de nouveaux signaux rassurants à l’Eular 2026
Laurinda Carré
L’étude JAK-POT collaboration : risque de cancer sous traitement par JAKi dans la PR
Pour rappel, l’étude ORAL surveillance avait conduit à plusieurs alertes de sécurité concernant un surrisque de cancer sous JAKi (1). Toutefois, cette étude ne portait que sur le tofacitinib et ne comparait pas les JAKi aux biomédicaments ayant d’autres mécanismes d’action (1). Par ailleurs, les études observationnelles, reflétant la pratique quotidienne ont rapporté des résultats discordants concernant ce risque. L’étude STAR-RA n’a pas mis en évidence d’augmentation du risque de cancer sous tofacitinib par rapport aux anti-TNF, tandis qu’une cohorte japonaise multicentrique a observé une incidence numériquement plus élevée du risque de cancer sous JAKi sans différence statistiquement significative après ajustement (2, 3).
L’étude JAK-POT
Cette étude internationale, réalisée à partir de 14 registres européens et québécois, a comparé le risque de cancer chez 37 559 patients atteints de polyarthrite rhumatoïde (PR) traités par JAKi, anti-TNFα, ou biomédicaments à autres mécanismes d’action.
Après ajustement sur les facteurs de confusion (notamment âge, sexe, IMC, caractéristiques de la PR, ligne de traitements/csDMARD/corticothérapie en cours, activité de la maladie, comorbidités), les JAKi n’étaient pas associés à une augmentation statistiquement significative du risque global de cancer (hors cancers cutanés non mélanocytaires), ni d’aucun sous-type de cancer par rapport aux anti-TNFα. En revanche, les biomédicaments à autres mécanismes d’action étaient associés à une incidence plus élevée de cancer global, de cancer pulmonaire et de cancer génital comparativement aux anti-TNFα, dans cette analyse.
En pratique
Cette large étude en vie réelle ne confirme pas le signal de surrisque de cancer observé dans ORAL SURVEILLANCE pour les JAKi. Bien qu’une augmentation numérique du risque global ait été observée, celle-ci n’était pas statistiquement significative. Ces résultats apparaissent globalement rassurants, mais nécessitent une confirmation par des études disposant d’un suivi plus prolongé avec un taux de survenue plus élevé pour les cancers rares.
• Ciribè B et al. Subtype-specific cancer risk with JAK inhibitors and biologic DMARDs in rheumatoid arthritis: comparative analysis within the JAK-POT collaboration. Eular 2026 : OP086.
Poursuite ou reprise d’un biomédicament après un cancer d’après le registre japonais NinJa
En pratique clinique, les traitements biologiques sont fréquemment interrompus lors de la survenue d’un cancer chez les patients atteints de PR, en raison de préoccupations concernant une possible altération de l’immunité antitumorale. Pourtant, de récentes études suggèrent que les biomédicaments, principalement les anti-TNFα, n’augmenteraient pas le risque de récidive ou de survenue d’un nouveau cancer chez des patients ayant un antécédent de cancer en rémission prolongée (4, 5).
Cette étude issue du registre national japonais NinJa (National Database of Rheumatic Diseases in Japan) a inclus 220 patients atteints de PR ayant présenté un cancer incident alors qu’ils étaient traités par un biomédicament, parmi plus de 15 000 patients suivis dans le registre. Au moment du diagnostic de cancer, 24 % des patients (n = 53) poursuivaient leur traitement biologique. Cette proportion atteignait 41 % chez ceux présentant un cancer de stade précoce (stades 0-1). Parmi les 167 patients ayant interrompu leur biomédicament, 33 % ont repris un traitement biologique après une médiane de 1,1 an. Plus de la moitié des patients atteints d’un cancer de stade intermédiaire (stades 2-3) a ainsi repris un biomédicament.
La poursuite du traitement dans les cancers de stade précoce et sa reprise dans les cancers de stade intermédiaire n’étaient pas associées à une diminution de la survie globale à 5 ans. Les principaux facteurs associés à la mortalité étaient l’âge, les cancers de stade IV et la corticothérapie ≥ 5 mg/j après le diagnostic de cancer.
En pratique
Cette étude suggère que, chez des patients sélectionnés, la poursuite d’un biomédicament après un cancer de stade précoce ou sa reprise après un cancer de stade intermédiaire pourrait être envisageable sans effet défavorable apparent sur la survie à moyen terme. Ces résultats doivent toutefois être interprétés avec prudence compte tenu du caractère observationnel de l’étude et du risque de biais de sélection.
• Siripongvutikorn Y et al. Safety in continuing and restarting biologics in rheumatoid arthritis patients with new-onset cancer – a retrospective multi-center study from the national database of rheumatic diseases in Japan. Eular 2026 : OP089.
Durée d’exposition aux traitements ciblés et risque de cancer dans les spondyloarthrites : données du SNDS
L’inflammation systémique chronique est reconnue comme un facteur de risque de cancer bien que des inquiétudes persistent quant au risque de malignité à long terme associé aux traitements immunomodulateurs (6). Alors que les thérapies ciblées sont largement utilisées dans les spondyloarthrites, les études observationnelles concernant l’effet de la durée cumulée d’exposition sur le risque de cancer restent limitées.
Les chercheurs de l’hôpital Henri-
Mondor (Créteil) ont inclus 56 591 patients à partir des données du système national des données de santé (SNDS, intégrant toutes les données de l’Assurance maladie), atteints de spondyloarthrite recevant des traitements ciblés (anti-TNFα, anti-IL-17, anti-IL-23 ou JAKi) entre 2014 et 2022.
Après un suivi médian de 5 ans, 1 224 cancers incidents ont été identifiés. Une exposition aux traitements ciblés supérieure à 6 mois par an était associée à une diminution du risque global de cancer par rapport à une exposition inférieure ou égale à 6 mois par an (HR pondéré 0,86 ; IC 95 % : 0,75-0,99). Cette association était principalement observée pour les hémopathies malignes (HR 0,65 ; IC 95 % : 0,42-0,99) sans réduction significative du risque de tumeurs solides.
En pratique
Cette large étude en vie réelle apporte des données rassurantes concernant la sécurité à long terme des traitements ciblés dans la spondylarthrite concernant le risque de cancer. L’une des hypothèses suggère qu’un meilleur contrôle de l’inflammation chronique pourrait contribuer à réduire le risque carcinologique. Ces résultats doivent néanmoins être interprétés avec prudence en raison du risque résiduel de facteurs de confusion inhérent aux études observationnelles.

Tankovic K et al. Risk of cancer according to duration of targeted therapy exposure in spondyloarthritis: a nationwide cohort study from the French health insurance database. Eular 2026 : OP0238.
Bibliographie
1. Ytterberg SR, Bhatt DL, Mikuls TR et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med 2022 ; 386 : 316‑26.
2. Khosrow-Khavar F, Desai RJ, Lee H et al. Tofacitinib and risk of malignancy: results from the safety of tofacitinib in routine care patients with rheumatoid arthritis (STAR-RA) study. Arthritis Rheumatol 2022 ; 74 : 1648‑59.
3. Uchida T, Iwamoto N, Fukui S et al. Comparison of risks of cancer, infection, and MACEs associated with JAK inhibitor and TNF inhibitor treatment: a multicentre cohort study. Rheumatology 2023 ; 62 : 3358‑65.
4. Raaschou P, Söderling J, Turesson C et al. Tumor necrosis factor inhibitors and cancer recurrence in swedish patients with rheumatoid arthritis: a nationwide population-based cohort study. Ann Intern Med 2018 ; 169 : 291‑9.
5. Xie W, Xiao S, Huang Y et al. A meta-analysis of biologic therapies on risk of new or recurrent cancer in patients with rheumatoid arthritis and a prior malignancy. Rheumatology 2020 ; 59 : 930‑9.
6. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008 ; 454 : 436‑44.
La PPR, star de l’Eular 2026 : confirmations en phase III des espoirs précédents
Baptiste Chevet
Longtemps restée orpheline de traitements ciblés malgré sa prévalence parmi les rhumatismes inflammatoires, la pseudo-polyarthrite rhizomélique (PPR) vit une véritable révolution. Après l’étude SEMAPHORE, en 2022, qui démontrait l’efficacité du tocilizumab pour diminuer l’activité de la maladie et la corticodépendance dans la PPR (1), le sarilumab a, l’année d’après, également montré son efficacité dans ces formes de PPR corticodépendantes (2). Depuis, la meilleure compréhension de sa physiopathologie, avec l’implication des voies JAK-STAT et de l’interleukine (IL)-17, ouvre de nouvelles voies thérapeutiques.
Cette édition de l’Eular confirme l’engouement autour de la PPR avec la présentation de deux essais de phase III positifs, un essai négatif, mais instructif, et les toutes premières recommandations internationales sur l’imagerie.
Le sécukinumab dans la PPR, résultats dithyrambiques de l’étude REPLENISH
L’aventure des traitements anti-IL-17 dans la PPR et l’artérite à cellules géantes (ACG) fait rêver tout chercheur : des données physiopathologiques relues 10 ans après leur publication (3, 4) font imaginer l’intérêt d’un anti-IL-17 dans l’ACG.
Le sécukinumab (SECU) a été évalué contre placebo dans l’ACG. Après l’étude TitAIN en phase II (5), l’essai de phase III, GCAPTAIN, ne remplit pas son objectif principal contre placebo (6). Toutefois, dans une analyse post-hoc de TitAIN, les patients ACG avec symptômes PPR étaient plus soulagés de leurs symptômes rhizoméliques s’ils recevaient du SECU plutôt qu’un placebo (7).
L’étude REPLENISH
Ainsi, l’essai REPLENISH, publié dans le New England Journal of Medicine le jour de sa présentation à l’Eular (8), évaluait le SECU contre placebo dans la PPR. Cet essai a inclus 381 patients atteints de PPR naïve de traitements ou corticodépendante. Les patients étaient répartis équitablement en trois groupes : SECU 150 mg, SECU 300 mg ou placebo. La corticothérapie était progressivement diminuée sur 24 semaines. Le critère de jugement principal était la rémission soutenue à la semaine 52, définie par une rémission à la semaine 12 sans aucune récidive PPR, ni diagnostic d’ACG nécessitant un traitement de secours.
Chacun des groupes était composé environ pour moitié de patients corticodépendants.
Les résultats
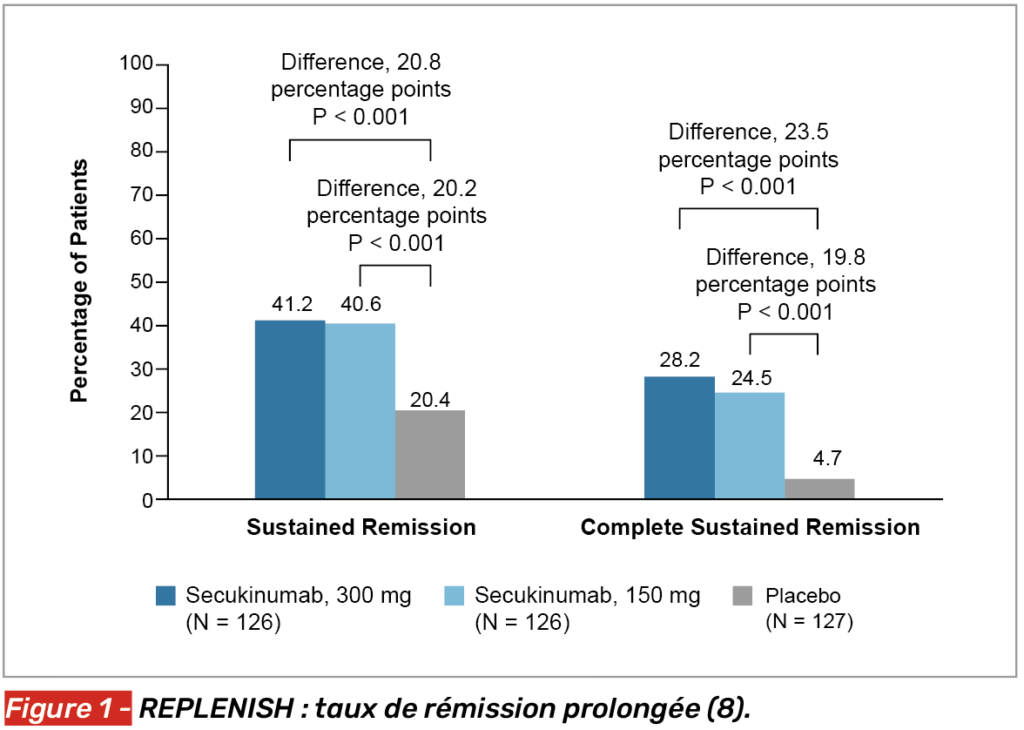
À S52, 41 % des patients des deux groupes SECU remplissaient le critère primaire, contre 20 % des patients sous placebo (p < 0,001) (Fig. 1). On notait également une épargne cortisonée, puisque l’on retrouvait une différence de près de 500 mg sur 1 an entre le groupe SECU 300 mg et le groupe placebo (SECU 300 : 1 603 mg, SECU 150 : 1 683 mg, placebo : 2 093 mg ; p < 0,001 vs SECU 300, p < 0,01).
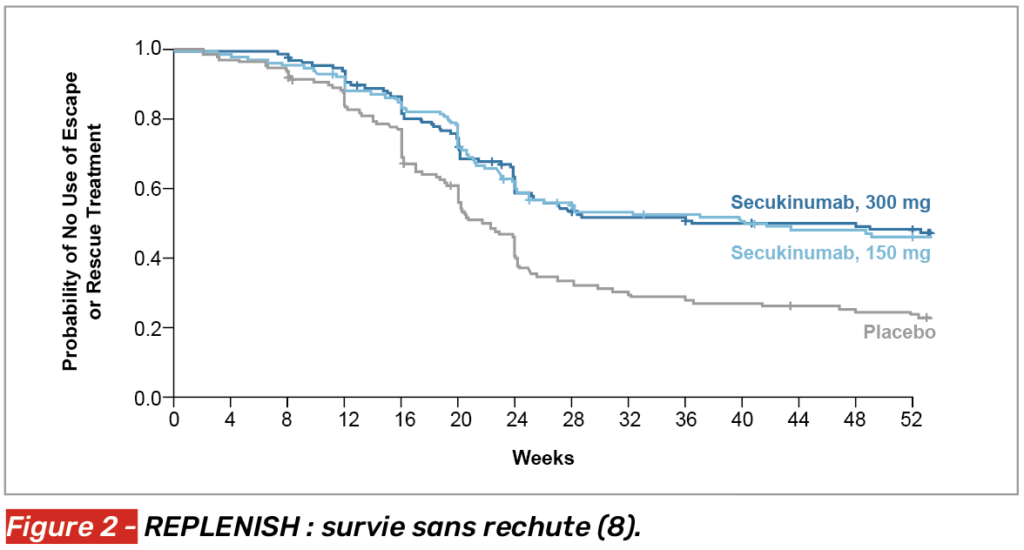
De plus, le risque de rechute ou de recours à un traitement de secours a été réduit de 50 % avec la dose de 300 mg (Fig. 2), prolongeant le délai avant la première rechute à 337 jours contre 157 dans le groupe placebo. Le profil de sécurité s’est révélé rassurant et comparable entre les groupes.
En pratique
Le sécukinumab devrait obtenir une place de choix dans l’arsenal thérapeutique de la PPR, à la fois dans sa forme naïve de traitement et corticodépendante. Face au tocilizumab, les anti-IL-17 présentent l’avantage majeur de ne pas éteindre artificiellement la production hépatique de CRP, permettant de continuer à suivre ce biomarqueur en routine.
De plus, bien que l’épargne cortisonée ne fût que de 500 mg sur 52 semaines, la diminution du risque de rechute avec le sécukinumab permettra d’éviter de nouvelles rechutes, et donc de multiples réascensions de la corticothérapie au cours du suivi du patient.
• Dejaco C et al. Secukinumab in polymyalgia rheumatica: results of the phase 3 REPLENISH trial. Eular 2026 : OP0116.
Baricitinib dans la PPR récente : résultats de l’essai JAK-SPARE
L’étude JAK-SPARE
Après le succès de l’essai français de phase II BACHELOR (9), l’étude académique autrichienne de phase III JAK-SPARE est venue évaluer l’efficacité du baricitinib, un inhibiteur oral de JAK, dans la PPR nouvellement découverte (moins de 3 semaines). Quarante-six patients dont le diagnostic datait de moins de 3 semaines et recevant une dose initiale de prednisone inférieure ou égale à 25 mg par jour ont été inclus. L’étude se décomposait en trois phases (Fig. 3). Les patients étaient randomisés 1:1 pour recevoir soit du baricitinib 4 mg, soit un placebo pendant 16 semaines, associés à un schéma de décroissance rapide des corticoïdes en 12 semaines. Ensuite, tous recevaient du baricitinib 4 mg des semaines 16 à 28. Les patients en rémission sans corticoïdes à S28 faisaient l’objet d’une nouvelle randomisation pour recevoir du baricitinib 4 mg jusqu’à S44, ou alors du baricitinib 2 mg pendant 8 semaines avant un arrêt. Le critère principal était la rémission sans corticoïdes à S16.
Vingt-trois patients recevaient du baricitinib, autant recevaient le placebo. Les femmes représentaient moins de la moitié de chaque groupe, l’âge moyen était de 65-66 ans. 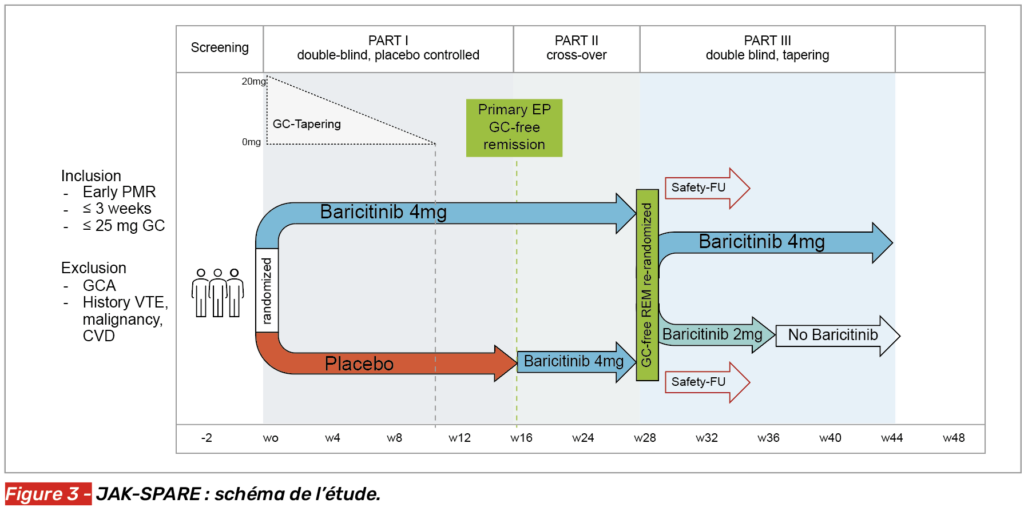
Les résultats
La rémission sans corticoïdes à S16 a été atteinte par 65 % des patients sous baricitinib contre seulement 17 % dans le groupe placebo (p < 0,01) (Fig. 4). Une différence comparable était déjà présente à S12. Les patients du groupe placebo présentaient une diminution de l’activité de la PPR après mise sous baricitinib dans la deuxième phase de l’étude, puisque 65 % d’entre eux étaient entrés en rémission sans corticoïdes à S28.
Le délai avant la première rechute était également significativement inférieur dans le groupe baricitinib. La dose cumulée de corticoïdes à la semaine 16 était significativement plus basse dans le bras baricitinib avec une médiane de 736 contre 1 065 mg dans le bras placebo (p = 0,015) (Fig. 5). Il est intéressant de noter que la dose médiane n’a pas changé entre S16 et S28 dans le groupe baricitinib, et était également très stable dans le groupe placebo, qui a reçu du baricitinib seulement de S16 à S28, augmentant seulement de 20 mg en 12 semaines.
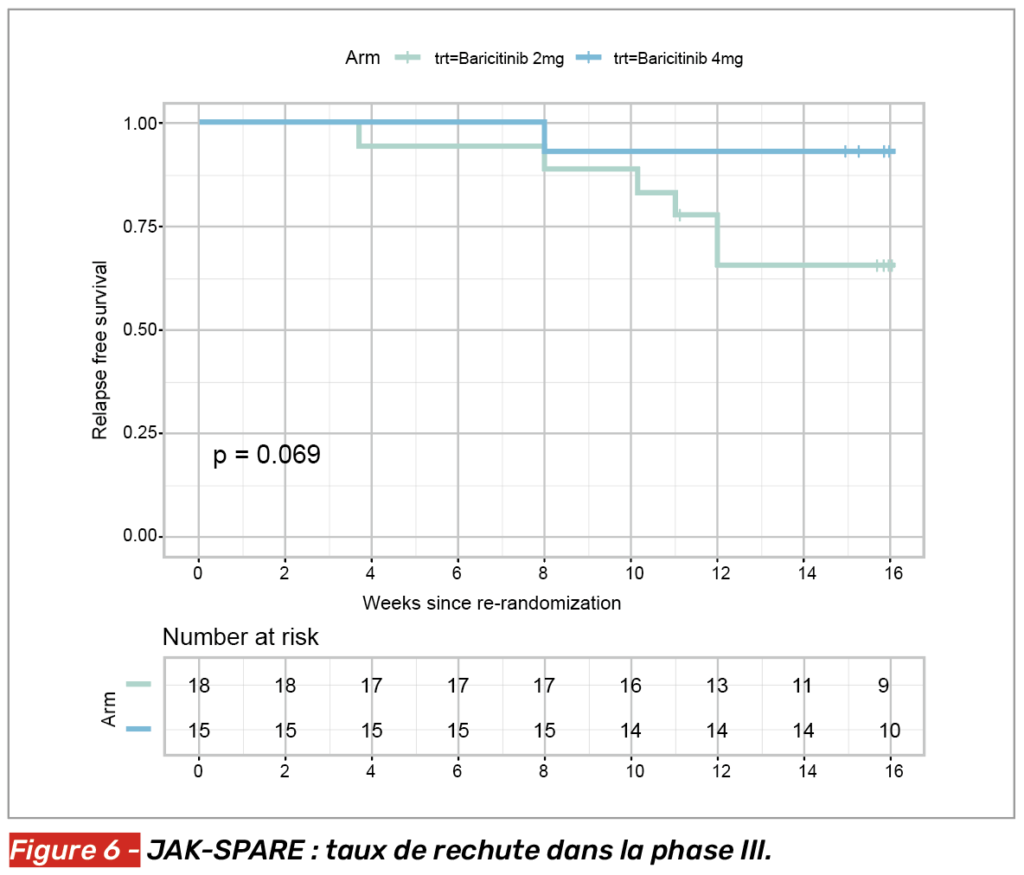
Notons que dans la phase III de l’étude, 33 patients en rémission sous baricitinib 4 mg ont été randomisés 1:1 pour soit continuer à recevoir le baricitinib à la même posologie (n = 18), soit entamer une décroissance avec 2 mg de baricitinib pendant 8 semaines avant un arrêt. Bien que la différence entre les deux groupes n’atteigne pas le seuil de significativité (possiblement dû au faible nombre de patients inclus dans cette analyse), les patients sous baricitinib 4 mg rechutaient moins que les autres (Fig. 6).
![]()
La tolérance
Elle s’est révélée similaire dans les deux groupes avec toutefois un cas d’adénocarcinome prostatique ayant été signalé à la semaine 18 sous baricitinib. À noter que les patients avec antécédents cardiovasculaires, thromboemboliques ou oncologiques n’avaient pas été inclus dans l’étude, et que moins de 10 % des patients de chaque groupe avaient déjà fumé dans sa vie.
En pratique
L’étude JAK-SPARE montre donc que baricitinib s’impose comme une option d’épargne cortisonique orale très efficace dès la phase initiale de la maladie. Cette étude associée aux récentes données à long terme rassurantes pour les JAKI présagent d’une nouvelle possibilité thérapeutique dans la PPR récente. Il reste à voir si l’étude JADORE-BARI, étude française incluant également des PPR corticodépendantes, conclura à l’efficacité du baricitinib également pour ces patients rechuteurs. Question relancée par cet essai JAK-SPARE et qui se pose désormais avec toutes les thérapies ciblées, la durée de traitement nécessaire pour éviter une rechute de la maladie est encore indéterminée, alors que la PPR est habituellement considérée comme curable.
• Lechner-Radner H et al. Baricitinib for remission induction and glucocorticoid sparing in new-onset polymyalgia rheumatica (JAK-SPARE): a phase III randomized controlled trial. Eular 2026 : LB0005.
Du rituximab dans la PPR récente : résultats moins favorables de l’essai REDUCE-PMR 1
Si les anti-IL-17 et les anti-JAK s’illustrent positivement, la place des anti-CD20 reste incertaine dans la PPR. Un essai pilote prometteur sur un faible effectif, BRIDGE-PMR, avait suggéré un intérêt potentiel (10), ce qui a conduit à la réalisation de l’essai de phase III néerlandais REDUCE-PMR 1 chez 116 patients ayant une PPR de diagnostic récent (moins de 3 mois). L’étude REDUCE-PMR 2, encore en cours, inclut des patients avec PPR corticodépendante.
L’étude RECUCE-PMR 1
Les patients recevaient une perfusion unique de rituximab 1 000 mg ou un placebo à l’inclusion, associée à un schéma de décroissance rapide de la prednisolone sur 17 semaines. Une nouvelle perfusion de rituximab 500 mg (ou placebo, selon le groupe) était autorisée à l’appréciation du clinicien après la 24e semaine.
Les résultats
Le critère principal, qui évaluait la rémission sans corticoïdes à la semaine 52 avec un score PMR-AS inférieur à 10, n’a pas atteint la significativité statistique, affichant 48,1 % dans le bras rituximab contre 34,6 % dans le bras placebo.
Même si l’ensemble des critères secondaires, tels que la rémission à S24, la dose cumulative de corticoïdes (1 761 mg en moyenne dans le groupe rituximab vs 2 295 mg du groupe placebo, à S52) et le délai de rémission penchaient numériquement en faveur du rituximab, aucune des analyses n’a atteint le seuil de significativité.
En pratique
L’effet produit s’avère trop modeste pour modifier nos pratiques cliniques actuelles.
• Kooijman N et al. Rituximab as a glucocorticoid-sparing therapy in newly diagnosed polymyalgia rheumatica: a randomized controlled trial (REDUCE PMR 1). Eular 2026 : OP0123.
Premières recommandations internationales pour l’utilisation de l’imagerie dans le diagnostic positif et le suivi de l’activité de la PPR
Parallèlement à ces avancées thérapeutiques, un groupe multidisciplinaire de 30 experts internationaux a formalisé les toutes premières recommandations concernant l’utilisation de l’imagerie comme l’échographie, l’IRM et la TEP-TDM dans la PPR. La principale force de ce travail est de conditionner l’intérêt de l’imagerie à la probabilité clinique pré-test du patient (Fig. 7).

Les éléments à retenir
• L’imagerie n’est pas recommandée en cas de probabilité faible pour exclure le diagnostic, ni en cas de probabilité élevée où la clinique et la biologie suffisent. Cette classification trouve sa valeur ajoutée en cas de probabilité intermédiaire ou face à une suspicion de diagnostics différentiels. Dans ce cas, des techniques d’imagerie à la recherche d’argument pour un diagnostic de PPR sont préconisées.
• À l’heure actuelle, aucune imagerie parmi l’échographie, l’IRM ou la TEP-TDM n’a montré de supériorité dans le diagnostic ou le suivi de l’activité de la PPR. Ainsi, le consortium considère que ces imageries doivent être complémentaires, et sans hiérarchie les unes par rapport aux autres, le choix de la modalité d’imagerie dépendant alors de l’expertise et de l’accès local.
• Un dépistage systématique d’une vascularite sous-clinique par imagerie vasculaire chez tout patient présentant une PPR isolée n’est pas validé par les données actuelles, et doit être réservé aux suspicions cliniques d’artérite à cellules géantes associée.
• De plus, lorsqu’elle est indiquée, l’imagerie doit idéalement être réalisée avant l’initiation de la corticothérapie ou avant une escalade thérapeutique en cas de rechute.
• Enfin, l’utilisation de l’imagerie pour le suivi de l’activité de la maladie ou pour prédire l’évolution clinique n’est pas recommandée, confirmant que l’évaluation clinique et biologique demeure le socle incontournable de la prise en charge de la PPR.
• Bond M et al. International recommendations for the use of imaging in the diagnosis and assessment of polymyalgia rheumatica. Eular 2026 : OP0118.
Les auteurs déclarent ne pas avoir de liens d’intérêt en rapport avec cet article.
Bibliographie
1. Devauchelle-Pensec V, Carvajal-Alegria G, Dernis E et al. Effect of tocilizumab on disease activity in patients with active polymyalgia rheumatica receiving glucocorticoid therapy: a randomized clinical trial. JAMA 2022 ; 328 : 1053-62.
2. Dasgupta B, Unizony S, Warrington KJ. Sarilumab in patients with relapsing polymyalgia rheumatica: a phase 3, multicenter, randomized, double blind, placebo controlled trial (SAPHYR). ACR Meeting 2022 ; LB0006.
3. Deng J, Younge BR, Olshen RA et al. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation 2010 ; 121 : 906-15.
4. Espígol-Frigolé G, Corbera-Bellata M, Planas-Rigol E et al. Increased IL-17A expression in temporal artery lesions is a predictor of sustained response to glucocorticoid treatment in patients with giant-cell arteritis. Ann Rheum Dis 2013 ; 72 : 1481-7.
5. Venhoff N, Schmidt WA, Bergner R et al. Safety and efficacy of secukinumab in patients with giant cell arteritis (TitAIN): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Rheumatol 2023 ; 5 : e341-50.
6. Stone JH, Venhoff N, Buttgereit F et al. Secukinumab for giant cell arteritis. NEJM Evid 2026 ; 5 : EVIDoa2600112.
7. Venhoff N, Schmidt W, Bergner R et al. Secukinumab in patients with giant cell arteritis with polymyalgia rheumatica symptoms: a post hoc analysis of the phase 2 TitAIN study. EULAR 2025 : OP0062.
8. Stone JH, Buttgereit F, Saraux A et al. Phase 3 trial of secukinumab in polymyalgia rheumatica. N Eng J Med 2026 ; Online ahead of print..
9. Clinical trials. University Hospital, Brest. BAriCitinib Healing Effect in earLy pOlymyalgia Rheumatica (BACHELOR Study). 2022. Disponible sur : clinicaltrials.gov/ct2/show/NCT04027101.
10. Marsman DE, den Broeder N, van den Hoogen FHJ et al. Efficacy of rituximab in patients with polymyalgia rheumatica: a double-blind, randomised, placebo-controlled, proof-of-concept trial. Lancet Rheum 2021 ; 3 : e758-66.