Résumé
La maladie de Still de l’adulte (MSA) est une maladie rare appartenant au spectre des maladies auto-inflammatoires. Son étiologie est encore mal connue. Elle est caractérisée par une triade clinique : fièvre prolongée, rash cutané maculeux saumoné évanescent et des arthralgies/arthrites dans un contexte de syndrome inflammatoire biologique. De nombreuses autres manifestations peuvent s’y associer comme une odynophagie, une splénomégalie, des adénopathies, une péricardite ou une pleurésie. Le diagnostic de MSA est un diagnostic d’élimination et repose sur des critères de classification parmi lesquels ceux de Yamaguchi et de Fautrel sont les plus utilisés chez l’adulte. Le pronostic global de la MSA est bon mais certaines complications, bien que rares, peuvent mettre en jeu le pronostic vital : le syndrome d’activation macrophagique, la CIVD, certaines hépatites fulminantes et complications pulmonaires ou cardiovasculaires. Au long cours, c’est d’une part, le pronostic fonctionnel qui sera concerné via les destructions articulaires et d’autre part, l’apparition d’une amylose AA secondaire à un mauvais contrôle de la maladie de fond qu’il faudra dépister. Le traitement a pour objectif la rémission sans corticoïde idéalement 3 mois après la première poussée ; il pourra être guidé par la présentation plutôt systémique ou articulaire pour choisir une biothérapie anticytokinique.
Abstract
Adult onset Still’s disease.
Adult onset Still’s disease (AOSD) is a rare disease in the spectrum of auto-inflammatory diseases. Its etiology is still poorly understood. It is characterized by a clinical triad: marked fever, evanescent salmon macular rash and arthralgia / arthritis and a biological inflammation. Odynophagia, splenomegaly, lymphadenopathy, pericarditis or pleurisy can also occur in AOSD. It is a diagnosis of elimination which could be relied on classification criteria of Yamaguchi and Fautrel. The overall prognosis for AOSD is good but some complications, although rare, can be life-threatening: macrophage activation syndrome, fulminant hepatitis and pulmonary or cardiovascular complications. In the long term, it is on the one hand the functional prognosis that will be impacted via joint destruction and on the other hand the appearance of AA amyloidosis secondary to poor control of the underlying disease that must be detected. The aim of treatment is remission without corticosteroids, ideally 3 months after the first attack; it may be guided by whether the presentation is systemic or articular in order to choose an anti-cytokine biotherapy.
La maladie de Still de l’adulte (MSA) est une affection rare dont la première description date du début des années 1970 par Bywaters (1). Ses mécanismes physiopathologiques exacts sont encore mal élucidés à ce jour mais l’immunité innée y tient un rôle primordial. Elle s’intègre dans la famille de maladies auto-inflammatoires et est polygénique (2). Sa présentation clinique est proche de la forme juvénile appelée arthrite juvénile idiopathique systémique. Dans ce travail, nous aborderons les caractéristiques épidémiologiques, cliniques, biologiques, diagnostiques, les complications et les principes de la prise en charge thérapeutique de la MSA.
Abréviations
• CAPS : syndrome périodique associé à la cryopyrine
• FMF : fièvre méditerranéenne familiale
• IL-1 : interleukine 1
• IL-6 : interleukine 6
• IV : intraveineuse
• JAK : Janus kinase
• LAI : leucémie angio-immunoblastique
• MSA : maladie de Still de l’adulte
• MVK : déficit en mévalonate kinase
• PNN : polynucléaires neutrophiles
• PVB19 : parvovirus B19
• SAM : syndrome d’activation macrophagique
• TRAPS : syndrome périodique associé au récepteur du TNF
• VEXAS : vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic
• VIH : virus de l’immunodéficience humaine
Épidémiologie
La MSA est une maladie rare sporadique, cosmopolite dont l’incidence annuelle varie entre 0,16 et 0,62 pour 100 000 individus dans le monde entier (3). Sa prévalence est estimée entre 0,73 et 6,77 pour 100 000 personnes (4). Des chiffres en augmentation dans les travaux les plus récents, attribués à une meilleure sensibilisation des médecins au diagnostic de MSA. Une discrète prédominance féminine est observée (3). Il s’agit d’une pathologie de l’adulte jeune avec un âge moyen au diagnostic de 38 ans [33,3-45,0] et une distribution selon deux pics : le premier entre 16 et 25 ans et le second entre 36 et 46 ans. Un retard diagnostic est cependant fréquent du fait de l’absence de signe pathognomonique et de l’hétérogénéité des symptômes possibles.
Clinique
Présentation classique (Fig. 1)
Une triade caractéristique définit la MSA : des pics fébriles > 39 °C associés à une éruption maculeuse ou maculopapuleuse saumonée et des arthralgies ou arthrites. La fièvre est le symptôme le plus fréquent, présent dans 84 à 100 % des cas selon les séries. Elle précède généralement les autres symptômes et s’accompagne d’une altération de l’état général. Elle est quotidienne, en pics élevés disparaissant en quelques heures au cours de la journée.
L’atteinte articulaire peut être retardée dans l’histoire naturelle de la MSA. Elle se rencontre dans 73 à 95 % des cas. Elle est localisée en début de maladie, touchant en particulier les genoux, les poignets, les chevilles, les coudes et les articulations interphalangiennes proximales.
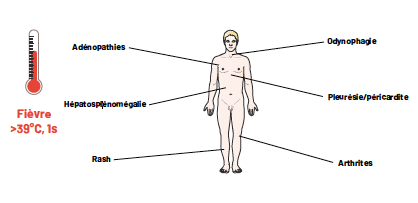
Figure 1 – Signes cliniques les plus fréquemment décrits dans la MSA.
Selon la forme de MSA, l’atteinte rhumatologique peut être au premier plan, devenir polyarticulaire puis se chroniciser. L’atteinte des deux poignets, sans atteinte des petites articulations des doigts est fréquente (Fig. 2). La présence de carpites fusionnantes bilatérales sans érosion des articulations métacarpophalangiennes ou des interphalangiennes proximales doit faire évoquer le diagnostic de MSA.
L’éruption cutanée décrite chez 68 à 81 % des patients est transitoire, non prurigineuse souvent concomitante des épisodes fébriles, présente en fin de journée et avec pour siège de prédilection la racine des membres, le tronc et les zones d’appui. L’histologie est aspécifique. L’éruption est parfois moins typique avec une présentation papuleuse ou urticarienne.
D’autres symptômes complètent cette triade clinique : une odynophagie ± pharyngite concomitante des épisodes fébriles, des adénomégalies, une hépatosplénomégalie, des myalgies et plus rarement des épanchements pleuraux ou péricardiques (3, 5).
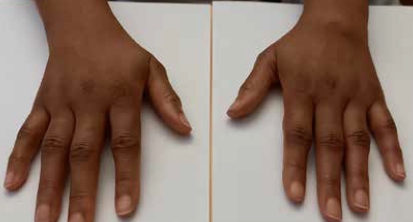
Figure 2 – Carpite associée à des arthrites de IPP et MCP chez une patiente en poussée de MSA.
Formes sévères
Des formes sévères de MSA engageant le pronostic vital du patient à court ou long terme existent et peuvent être un mode d’entrée dans la maladie. Le syndrome d’activation macrophagique (SAM), décrit chez 12 à 14 % des patients selon les séries, est l’une des complications les plus craintes avec une mortalité rapportée entre 10 et 20 %. Sa présentation possède des similarités avec une poussée de MSA : fièvre, splénomégalie, AEG, etc. Certaines atypies doivent cependant alerter le clinicien : une fièvre persistante, l’absence d’hyperleucocytose ou des cytopénies et une hypofibrinogénémie. Une coagulation intravasculaire disséminée peut être associée au SAM ou isolée dans de rares cas tout comme les micro-angiopathies thrombotiques. Dans la MSA, les perturbations du bilan hépatique sont fréquentes et de rares cas d’hépatites fulminantes sont décrits. Des tableaux de tamponnade ou de myocardite peuvent compliquer les péricardites, plus classiques dans la MSA. Les complications pulmonaires sont rares mais existent : de l’hypertension artérielle pulmonaire, une détresse respiratoire aiguë ou encore un tableau de pneumopathie interstitielle ont été rapportés dans quelques cas (6, 7).
Biologie
La MSA en poussée s’accompagne d’un syndrome inflammatoire biologique, parfois intense, avec une élévation de la CRP, une hyperleucocytose à polynucléaires neutrophiles, une hyperferritinémie marquée ainsi qu’une diminution nette de sa fraction glycosylée (< 20 %). Une cytolyse hépatique est fréquente. Des travaux rapportent l’utilisation des dosages sériques cytokiniques d’IL-1, d’IL-6, d’IL-18, ainsi que de certaines familles des protéines S100 pour le diagnostic de MSA mais ils ne sont pas encore recommandés en pratique courante. Les examens immunologiques ne montrent pas de signe d’auto-immunité (8).
Critères diagnostiques
Il n’existe pas de manifestation clinique ou paraclinique pathognomonique de MSA. Sa présentation très hétérogène rend son diagnostic complexe et souvent retardé. Il s’agit d’un diagnostic d’exclusion reposant sur des critères diagnostiques. Les critères les plus validés et utilisés en pratique courante sont ceux de Yamaguchi et de Fautrel (9, 10) (Fig. 3).
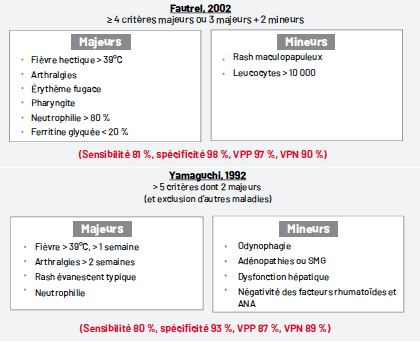
Figure 3 – Critères diagnostiques de MSA.
Diagnostics différentiels
Le clinicien doit avant de retenir un diagnostic de MSA s’employer à en éliminer les diagnostics différentiels tant par l’anamnèse que la clinique ou grâce à des examens paracliniques orientés. Les principaux diagnostics différentiels usuellement rapportés sont illustrés dans la figure 4 (4, 11).

Figure 4 – Diagnostics différentiels de la MSA.
L’apparition de la MSA après l’âge de 60 ans est rare dans la littérature. Dans certaines séries cette forme représente 7-10 % des patients (3, 12).
En 2020, une nouvelle maladie appelée le VEXAS syndrome a été décrite. Il s’agit d’une maladie auto-inflammatoire liée à la mutation somatique du gène UBA1, identifiée chez des hommes seniors présentant des signes communs avec la MSA. Elle peut se manifester par un syndrome inflammatoire fébrile, une anémie macrocytaire, des signes cutanés variés, des arthralgies ou arthrites (13). Le VEXAS syndrome est un nouveau diagnostic différentiel de la MSA et, en 2022, tout patient de plus de 60 ans suspect de MSA devrait bénéficier d’une recherche de néoplasie myéloïde et de mutation UBA1 en Sanger (Fig. 5).

Figure 5 – Le syndrome VEXAS.
Traitement
La MSA et sa forme d’apparition plus précoce, l’arthrite juvénile idiopathique, reposent sur les mêmes principes thérapeutiques. Par sa rareté et son hétérogénéité la MSA manque encore de recommandations précises et d’essais prospectifs multicentriques (2). En cas de suspicion de diagnostic de MSA et dans l’attente d’une confirmation, le traitement est essentiellement symptomatique et adapté à la présentation clinique, le plus souvent des AINS sont proposés. La corticothérapie générale à forte dose reste le traitement de première intention, avec l’utilisation initiale de la voie IV en cas d’atteinte viscérale grave. Elle peut être associée d’emblée dans de rares cas à un anti-IL-1, après réunion de concertation pluridisciplinaire (RCP) ou avis auprès des centres de référence en France. Actuellement, l’objectif est d’atteindre la dose de 0,1 mg/kg/j de corticoïdes à 6 semaines et l’arrêt complet à 3 mois. À défaut, le patient doit être considéré en réponse inadéquate et un traitement de fond doit être envisagé (14). Dans les formes systémiques, on préférera une biothérapie par anti-IL1 aux anti-IL6. Ces biothérapies peuvent être utilisées en monothérapie. Dans les formes articulaires ou paucisymptomatiques le méthotrexate est une option thérapeutique. D’autres traitements comme les anti-TNF, la ciclosporine A ou les anti-JAK peuvent être proposés en cas d’échec des thérapeutiques de première intention (14). Dans la phase d’entretien en cas de rémission prolongée (> 6 mois), une décroissance des traitements de fond est proposée par réduction ou espacement des doses.
Évolution et pronostic
L’histoire naturelle de la MSA se décrit selon trois modèles distincts :
- la forme monocyclique définie par un épisode systémique unique sans récidive,
- la forme polycyclique ou intermittente associant des poussées multiples systémiques ou articulaires dont la sévérité évoluera decrescendo dans le temps séparées par des épisodes de rémission,
- la forme chronique avec une activité persistante dans le temps généralement associée à une polyarthrite avec un risque de handicap par destruction articulaire important.
La MSA est une maladie au pronostic habituellement favorable mais certaines manifestations rares ou complications au long cours comme l’amylose AA peuvent mettre en jeu le pronostic vital à court ou long terme du patient (15).
Conclusion
La MSA est une maladie rare de diagnostic difficile dans lequel le rhumatologue joue souvent un rôle clé. Son diagnostic peu aisé peut entraîner un retard de diagnostic important à l’origine d’un délai de prise en charge thérapeutique et dans certains cas du développement de certaines complications pouvant affecter le pronostic vital mais aussi fonctionnel du patient.
Les auteurs déclarent ne pas avoir de liens d’intérêts pour ce travail.
Bibliographie
1. Bywaters EG. Still’s disease in the adult. Ann Rheum Dis 1971 ; 30 : 121‑33.
2. Tomaras S, Goetzke CC, Kallinich T, Feist E. Adult-Onset Still’s Disease: Clinical Aspects and Therapeutic Approach. J Clin Med 2021 ; 10 : 733.
3. Efthimiou P, Kontzias A, Hur Pet al. Adult-onset Still’s disease in focus: Clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum 2021 ; 51 : 858‑74.
4. Mitrovic S, Fautrel B. Still ou Pseudo-Still : difficultés et pièges du diagnostic de maladie de Still de l’adulte. Revue du rhumatisme 2018 ; 85 : 259‑66.
5. Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still’s disease. J Autoimmun 2018 ; 93 : 24‑36.
6. Fauter M, Gerfaud-Valentin M, Delplanque M et al. Adult-onset Still’s disease complications. Rev Med Int 2020 ; 41 : 168‑79.
7. Mitrovic S, Fautrel B. Complications of adult-onset Still’s disease and their management. Expert Rev Clin Immunol 2018 ; 14 : 351‑65.
8. Mitrovic S, Fautrel B. New Markers for Adult-Onset Still’s Disease. Joint Bone Spine 2018 ; 85 : 285‑93.
9. Yamaguchi M, Ohta A, Tsunematsu T et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol 1992 ; 19 : 424‑30.
10. Fautrel B, Zing E, Golmard J-L et al. Proposal for a new set of classification criteria for adult-onset still disease. Medicine 2002 ; 81 : 194‑200.
11. Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol 2008 ; 22 : 773‑92.
12. Mollaeian A, Chen J, Chan NN et al. Adult onset Still’s disease in the elderly: a case-based literature review. BMC Rheumatol 2021 ; 5 : 12.
13. Georgin-Lavialle S, Terrier B, Guedon AF et al. Further characterization of clinical and laboratory features occurring in VEXAS syndrome in a large-scale analysis of multicenter case-series of 116 French patients. Br J Dermatol 2021; doi: 10.1111/bjd.20805.
14. Fautrel B. Protocole national de diagnostic et de soins PNDS 2017 Maladie de STILL de l’adulte et de la forme systémique de l’arthrite juvénile idiopathique ayant évolué jusqu’à l’âge adulte [Internet]. 2017. Disponible sur : www.has-sante.fr/upload/docs/application/pdf/2018-08/pnds_still_de_ladulte_vfinale_2.pdf
15. Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-onset Still’s disease. Autoimmun Rev 2014 ; 13 : 708‑22.