L’essentiel en un clin d’œil
• Modulation neuro-immunitaire chez des patients atteints de PR présentant une réponse insuffisante ou une intolérance aux DMARD biologiques ou synthétiques ciblés : résultats à 12 et 24 semaines d’une étude randomisée, contrôlée par placebo simulé, en double aveugle (étude RESET-RA).
• Inhibition double de JAK/ROCK dans la polyarthrite rhumatoïde : résultats d’une étude de phase II sur le CPL’116.
• Espacement de l’anti-TNF ou réduction de la dose de méthotrexate chez les patients atteints de polyarthrite rhumatoïde en rémission ou en faible niveau d’activité : l’essai SORAIRO.
• Abatacept chez les individus à risque de développer une polyarthrite rhumatoïde : résultats de l’étude sur la prévention de l’arthrite dans la phase pré-clinique de la polyarthrite rhumatoïde avec les résultats à long terme de l’abatacept (étude ALTO).
• Effets de l’abatacept versus l’hydroxychloroquine sur la progression vers la polyarthrite rhumatoïde chez les patients atteints de rhumatisme palindromique : essai clinique randomisé multicentrique de 2 ans (étude PALABA).
TOP 1 – Modulation neuro-immunitaire en cas de réponse insuffisante ou d’intolérance aux b/tsDMARDs : résultats à 12 et 24 semaines de RESET-RA
L’activation du réflexe neuro-immunitaire médié par le nerf vague par stimulation électrique peut réduire la production de plusieurs cytokines inflammatoires, offrant ainsi une nouvelle approche thérapeutique pour la gestion des maladies auto-immunes comme la polyarthrite rhumatoïde (PR). La stimulation du nerf vague augmente également les médiateurs spécialisés de résolution de l’inflammation, favorisant la résolution de l’inflammation par plusieurs mécanismes, notamment en augmentant le nettoyage des débris cellulaires et la minéralisation osseuse. L’activation de ce réflexe par une stimulation électrique de 60 secondes inhibe des voies intracellulaires, entraînant une suppression de 30 à 70 % de plusieurs cytokines pro-inflammatoires, avec une réduction soutenue jusqu’à 24-48 heures après stimulation.
L’étude RESET-RA
L’objectif de l’essai RESET-RA était d’évaluer l’efficacité et la tolérance d’un dispositif implantable de neurostimulation cervicale pour le traitement des adultes atteints de PR modérément à sévèrement active, ayant une réponse insuffisante ou une intolérance à au moins un traitement préalable par DMARD biologique (bDMARD) ou DMARD synthétique ciblé (tsDMARD).
Cette étude randomisée, en double aveugle, contrôlée par placebo simulé, a recruté 242 patients dans 41 centres aux États-Unis. Tous sont restés sous DMARDs conventionnels stables et ont interrompu leur b/tsDMARD avant la procédure d’implantation. Les patients ont été randomisés selon un ratio 1:1 pour recevoir une stimulation active (traitement) ou non active (contrôle).
Le critère principal était la proportion de patients atteignant une réponse ACR20 à la semaine 12 par rapport à la valeur de référence au jour du consentement éclairé, avant l’implantation. Les critères secondaires et exploratoires comprenaient le taux de réponse modérée/bonne selon le DAS28-CRP et Eular, les taux de faible activité maladie (LDA) et de rémission, ainsi que la proportion de sujets avec progression des érosions (> 0,5 d’augmentation du score d’érosion) mesurée par IRM (RAMRIS).
Les patients nécessitant un traitement additionnel avant la semaine 12 ou pour lesquels les données ACR étaient manquantes à cette date étaient considérés comme non-répondeurs. Après la semaine 12, l’étude est devenue en ouvert avec un passage en cross-over du groupe contrôle vers la stimulation active, et une nouvelle évaluation d’efficacité était effectuée à la semaine 24. L’augmentation du traitement de la PR après la semaine 12 était laissée à la discrétion de l’investigateur.
Les patients
Les caractéristiques démographiques et cliniques initiales étaient bien équilibrées et représentatives de la population PR aux États-Unis : âge moyen 56 ans, 86 % de femmes, durée moyenne de PR 12 ans, IMC 30, 15 articulations douloureuses et 10 articulations gonflées (sur 28 articulations évaluées). En moyenne, les patients avaient une réponse insuffisante ou une intolérance à 2,6 traitements b/tsDMARDs avant l’inclusion, avec 40 % ayant échoué à un traitement, 21 % à deux traitements, et 39 % à trois traitements ou plus. En outre, 53 % étaient séropositifs (FR et/ou anti-CCP) et la CRP ultrasensible moyenne était de 8,2 mg/l.
Résultats
La réponse ACR20 à la semaine 12 montrait une différence statistiquement significative entre les groupes traitement et contrôle (p = 0,0209 ; IC 95 % = 0,6-23,1) (Fig. 1). À la semaine 24, la réponse ACR20 a augmenté après cross-over, atteignant 51,5 % dans le groupe traitement et 53,1 % dans le groupe contrôle devenu actif (Fig. 1). Pour les patients avec une seule exposition préalable à un bDMARD (analyse pré-spécifiée), la réponse ACR20 à la semaine 12 était plus élevée : 45,5 % pour le traitement contre 19,0 % pour le contrôle (p = 0,0037 ; IC 95 % = 8,6-44,3).
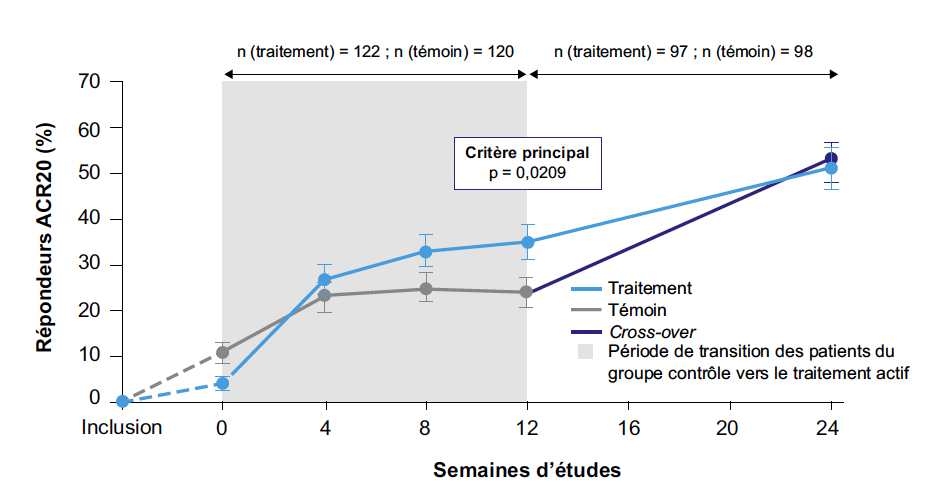
Figure 1 – RESET-RA : pourcentages de répondeurs ACR20 aux semaines 12 et 24 dans les groupes traités par stimulation vagale active (traitement) et non active (placebo).
Tous les critères secondaires et plusieurs exploratoires penchaient en faveur du groupe traitement. La proportion de patients avec une réponse modérée à bonne selon DAS28-CRP et Eular à la semaine 12 était significativement plus élevée dans le groupe traitement (60,7 %) que dans le groupe contrôle (41,7 %) (p = 0,0012 ; IC 95 % = 7,3-31,7). Les taux de faible activité de la maladie et de rémission au DAS28-CRP à la semaine 12 étaient également significativement supérieurs dans le groupe traitement et s’amélioraient jusqu’à la semaine 24. La proportion de patients avec progression des érosions selon RAMRIS était inférieure dans le groupe traitement par rapport au groupe contrôle. À la semaine 24, 81 % des patients n’avaient pas reçu de doses élevées supplémentaires de corticoïdes ou de b/tsDMARDs.
Tolérance
L’implantation et la stimulation étaient globalement sûres et bien tolérées, tant en phase aiguë qu’en suivi prolongé. Le taux d’effets indésirables graves liés à l’étude était faible (1,7 %). L’effet indésirable le plus fréquent lié au traitement était une dysphonie légère à modérée ou un dysfonctionnement des cordes vocales. Aucun décès n’a été rapporté.
En pratique
La modulation neuro-immunitaire via la stimulation du nerf vague constitue une nouvelle modalité thérapeutique potentielle pour les patients atteints de PR ayant échoué à au moins un bDMARD ou tsDMARD. L’étude RESET-RA a atteint son objectif avec une amélioration significative chez les patients stimulés activement. La procédure d’implantation, le dispositif et la stimulation étaient bien tolérés avec des taux faibles et attendus d’effets indésirables, sans cas liés d’infections graves, d’événements cardiovasculaires majeurs ou de cancers. L’activation des voies neuronales grâce à ce neurostimulateur implantable peut réguler l’inflammation et offrir potentiellement une alternative aux traitements pharmaceutiques actuellement disponibles pour la PR.
• Tesser J, Crowley A, Wickersham P et al. Neuroimmune modulation for treatment of rheumatoid arthritis in adults with inadequate response or intolerance to biological or targeted synthetic DMARDs: results at 12 and 24 weeks from a randomized, sham-controlled, double-blind study (RESET-RA study). Eular 2025 ; OP0190.
TOP 2 – Inhibition double de JAK/ROCK : résultats d’une étude de phase II sur le CPL’116
Les inhibiteurs de JAK (JAKi) sont sous surveillance depuis l’étude ORAL Surveillance, en raison du risque cardiovasculaire augmenté sous tofacitinib en comparaison aux anti-TNF. Le CPL’116 est une molécule qui agit par une inhibition sélective duale des kinases JAK et des kinases Rho (ROCK) combinant les effets anti-inflammatoires des JAKi avec les effets anti-fibrotiques et cardioprotecteurs des ROCK. Cette double inhibition vise notamment à réduire la dysfonction endothéliale, un mécanisme clé dans la morbi-mortalité cardiovasculaire associée à la PR.
L’étude
Dans cet essai de phase II, randomisée, en double aveugle, contrôlée par placebo, en groupes parallèles, d’une durée de 12 semaines, l’efficacité et la tolérance de CPL’116 ont été évaluées chez des patients atteints de PR modérée à sévère en réponse inadéquate au méthotrexate (MTX). Ils devaient donc présenter une maladie modérée à sévère avec une réponse inadéquate au méthotrexate (15-25 mg/sem) définie par un DAS28 > 3,2, au moins 6/68 articulations douloureuses et gonflées et une CRP > 7 mg/l.
Après avoir complété toutes les évaluations de dépistage, 106 patients ont été randomisés selon un ratio 1:1:1:1 dans chacun des quatre groupes de traitement suivant :
• CPL’116 60 mg (n = 27),
• CPL’116 120 mg (n = 25),
• CPL’116 240 mg (n = 26)
• ou placebo (n = 28),
administrés par voie orale deux fois par jour pendant une durée de 12 semaines. L’utilisation concomitante des corticoïdes oraux était autorisée jusqu’à 10 mg/j.
Le critère de jugement principal était la variation du DAS28 entre l’inclusion et la 12e semaine. Les patients inclus étaient des femmes (75 %), un âge moyen de 54,4 ans (± 10,5).
Résultats
À 12 semaines, le CPL’116 a entraîné une réduction du score DAS28-CRP de manière dose-dépendante, avec une amélioration statistiquement significative à la dose de 240 mg x 2/j (-0,887 ; p = 0,01). Pour les autres posologies, la différence n’était pas significative (Fig. 2).
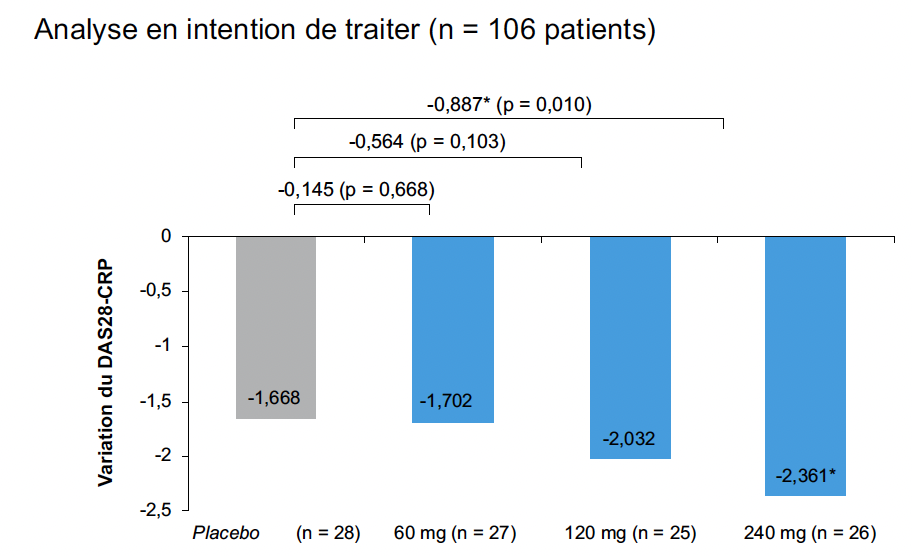
Figure 2 – Variation du DAS28-CRP entre l’inclusion et la semaine 12 dans les groupes CPL’116 (60, 120 et 240 mg) et placebo.
Tolérance
Trois participants (2,8 %) ont interrompu définitivement le traitement en raison d’effets indésirables. Aucun effet indésirable n’a conduit à une réduction de dose ni à un décès. Dans l’analyse de tolérance, les paramètres lipidiques ont également été évalués et comparés aux autres JAKi montrant un profil favorable pour le CPL’116 avec une absence d’élévation du LDL-
cholestérol et des triglycérides. Notamment, le CPL’116 est à ce jour le seul JAKi présentant un tel profil lipidique favorable.
En pratique
Cette nouvelle molécule pourrait devenir une option thérapeutique efficace dans la PR en échec du MTX. Son profil de tolérance semble plus favorable que les autres JAKi sur une durée de suivi limitée à 16 semaines, en particulier sur le profil lipidique et il faudra maintenant le confirmer sur une période plus prolongée. CPL’116 pourrait être un traitement prometteur pour les patients atteints de PR et d’autres maladies auto-immunes, en particulier celles à composante fibrotique, comme la PR-ILD (PR avec atteinte pulmonaire interstitielle) ou la fibrose pulmonaire idiopathique (IPF).
• Wieczorek M, Kisiel B, Włodarczyk D et al. Dual JAK/ROCK inhibition in rheumatoid arthritis – Results of a phase 2 study of CPL’116. Eular 2025 ; OP0193.
TOP 3 – Espacement de l’anti-TNF ou réduction de la dose de méthotrexate chez les patients en rémission ou en faible niveau d’activité
La décroissance thérapeutique dans la PR contrôlée sous traitement associant csDMARDs et biothérapies ciblées est une stratégie qui expose à une rechute du rhumatisme. L’étude TARA n’avait pas mis en évidence de différence significative entre la réduction de posologie du MTX ou celle de l’anti-TNF.
Dans ce protocole, la décroissance était conduite de manière relativement rapide : réduction à la moitié, puis aux trois quarts de la dose, puis arrêt complet tous les 3 mois si la rémission se maintenait – en commençant dans un bras par le MTX et dans l’autre par l’anti-TNF.
L’étude SORAIRO
L’objectif de l’essai présenté ici était d’évaluer une stratégie de décroissance progressive fondée soit sur l’espacement d’un nouvel inhibiteur du TNF, l’ozoralizumab (OZR), soit sur une diminution plus lente de la posologie du MTX. L’OZR est un nanobody trivalent innovant, disponible au Japon, administré à la posologie de 30 mg/mois. Il se distingue des anticorps classiques par l’absence de fragment Fc, remplacé par une région de liaison à l’albumine humaine. Cette caractéristique confère à la molécule une demi-vie prolongée en limitant son épuration rénale, tout en maintenant une forte affinité pour sa cible.
SORAIRO est un essai multicentrique, ouvert, randomisé, en groupe parallèle, de non-infériorité ayant inclus des patients en rémission (CDAI ≤ 2,8) ou faible activité (CDAI ≤ 10) après 3–4 ans de traitements combinés OZR + MTX. Trois bras ont été comparés :
• traitement continu (groupe de référence, n = 46),
• espacement de l’OZR (toutes les 4→ 6 → 8 semaines, n = 48),
• réduction de la dose de MTX (réduction de 2 mg/semaine toutes les 8 semaines, n = 44).
Le critère de jugement principal était la proportion de patients maintenant un CDAI ≤ 10 à la semaine 48. Ont également été évaluées les proportions de patients sans MTX dans le groupe réduction de dose MTX et ceux atteignant un intervalle de 8 semaines pour l’OZR dans le groupe espacement OZR. D’autres indices d’efficacité et la sécurité ont aussi été étudiés.
Résultats
Au total, 144 patients ont été inclus dans l’analyse en per-protocole. L’âge moyen était de 58,2 ans et 75 % étaient des femmes. Quatre-vingt-neuf patients (62 %) étaient en rémission CDAI (≤ 2,8). À l’issue des 48 semaines de suivi, la cible thérapeutique correspondant à un CDAI ≤ 10 était atteinte chez 98 % des patients du groupe en traitement continu, contre 79 % dans le groupe bénéficiant d’un espacement des injections d’OZR et 73 % chez ceux soumis à une réduction progressive du MTX. Toutefois, aucune des deux stratégies de décroissance n’a permis de démontrer la non-infériorité par rapport au maintien du traitement complet, le seuil de non-infériorité ayant été fixé à -18 % (Fig. 3). Pour les patients en rémission CDAI (≤ 2,8) au début de la décroissance thérapeutique, le CDAI ≤ 10 était maintenu chez 100 % du groupe poursuite, 93 % du groupe espacement OZR et 92 % du groupe réduction MTX, avec une rémission maintenue de façon comparable entre les groupes (77-79 %). Un statut sans MTX a été atteint chez 49 % des patients du groupe réduction MTX, sans facteurs prédictifs significatifs identifiés. En revanche, 80 % des patients du groupe espacement OZR ont atteint un intervalle de 8 semaines pour la posologie d’OZR, ce qui était associé à la rémission CDAI au départ (OR = 10,182 ; p = 0,007) et à un taux d’albumine > 38 g/l (OR = 5,500 ; p = 0,027). Parmi les patients ayant connu des poussées, 89 % dans le groupe espacement OZR et 83 % dans le groupe réduction MTX ont retrouvé un CDAI ≤ 10 après un traitement de rattrapage (augmentation des doses initiales d’OZR ou de MTX). La variation moyenne du score total Sharp modifié (STSm) sur 48 semaines indiquait une rémission structurale (delta score STSm < 0,5) dans tous les groupes.
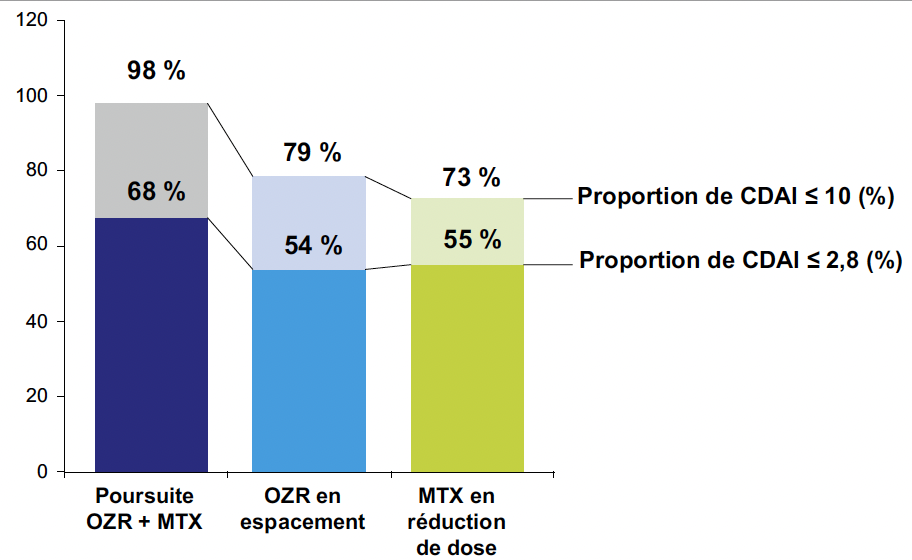
Figure 3 – SORAIRO : pourcentages de patients en rémission (CDAI ≤ 2,8) ou en faible niveau d’activité (CDAI ≤ 10) à la semaine 48 dans les groupes décroissance du méthotrexate (MTX) ou de l’ozoralizumab (OZR) et dans le groupe maintien thérapeutique MTX/OZR.
Tolérance
Des événements indésirables ont été observés chez 68 % des patients du groupe poursuite, 59 % du groupe espacement OZR et 58 % du groupe réduction MTX. Deux événements indésirables graves sont survenus dans chaque groupe.
En pratique
Chez les patients ayant atteint la rémission avec une association anti-TNF et MTX, l’espacement de l’anti-TNF ou la réduction de dose de MTX peuvent être des options envisageables et favorables sous surveillance clinique attentive. Cette étude souligne également l’importance d’atteindre la rémission pour réussir la décroissance thérapeutique en particulier pour l’espacement de l’anti-TNF.
• Imai Y, Atsumi T, Ishii T et al. Spacing of a TNF blocker and dose reduction of methotrexate in patients with rheumatoid arthritis in remission or low disease activity with ozoralizumab and methotrexate: a randomised, controlled trial (SORAIRO trial). Eular 2025 ; LB0006.
TOP 4 – Résultats de l’étude sur la prévention de l’arthrite dans la phase pré-clinique avec les résultats à long terme de l’abatacept
Plusieurs essais cliniques randomisés visant à retarder ou à prévenir l’apparition de la PR chez des individus à risque ont été publiés, avec des résultats variables. Dans l’étude APIPPRA, il a été observé que 52 semaines de traitement par abatacept (ABA) réduisaient les taux de progression vers la PR définis par le développement de synovite clinique dans ≥ trois articulations, ou le diagnostic de PR selon les critères ACR/Eular 2010, avec des effets qui se maintenaient au-delà de la période de traitement. En revanche, il existe moins de données sur les effets à long terme sur la prévention d’une PR.
L’étude ALTO
APIPPRA était un essai de phase IIB, randomisé, en double aveugle, contrôlé par placebo, ayant recruté 213 individus ACPA+ FR+ ou ACPA élevé FR- présentant des arthralgies inflammatoires sans synovite clinique. Les participants ont été randomisés pour recevoir 52 injections sous-
cutanées hebdomadaires de placebo (n = 103) ou d’ABA à 125 mg (n = 110), et ont été suivis au moins pendant 52 semaines supplémentaires après l’arrêt du traitement. Tous les participants à l’étude APIPPRA ont été invités à participer à l’étude d’extension ALTO.
Pour évaluer les événements survenus entre la fin d’APIPPRA et le début d’ALTO, ainsi que pendant le suivi à plus long terme, le délai avant le premier traitement par médicaments antirhumatismaux modificateurs de la maladie (hors corticostéroïdes) a été inclus dans le critère d’évaluation principal si cet événement survenait en premier. Les critères d’évaluation principaux ont également été analysés selon la stratification fondée sur les auto-anticorps sériques définis lors de la visite initiale de l’étude APIPPRA. Pour évaluer la tolérance, les événements indésirables graves ont été recensés, ainsi que les événements d’intérêt particulier, en mettant l’accent sur les épisodes cardiovasculaires, cancéreux et infectieux.
Les patients
Parmi les 213 patients randomisés dans APIPPRA, 143 ont été inclus dans ALTO (71 dans le groupe ABA et 72 dans le groupe placebo). Les caractéristiques des patients étaient comparables à celles de l’étude APIPPRA : âge moyen 48 ans, 78 % de femmes et 60 % étaient des fumeurs actifs ou anciens.
Résultats
La durée médiane de suivi depuis la randomisation dans l’étude APIPPRA était de 66 mois (Q25-Q75 : 54-78). À la fin du suivi, un total de 119 événements correspondant au critère principal a été enregistré.
• À la fin de la première année suivant la randomisation dans APIPPRA, la proportion cumulée de participants atteints d’arthrite était de 29 % dans le groupe placebo et de 6 % dans le groupe ABA (différence de 23 %).
• À la fin de la deuxième année, ces taux étaient respectivement de 40 et 30 % (différence de 10 %).
• À la fin de la troisième et de la quatrième années, les différences étaient de 4 et 2 % respectivement (Fig. 4).
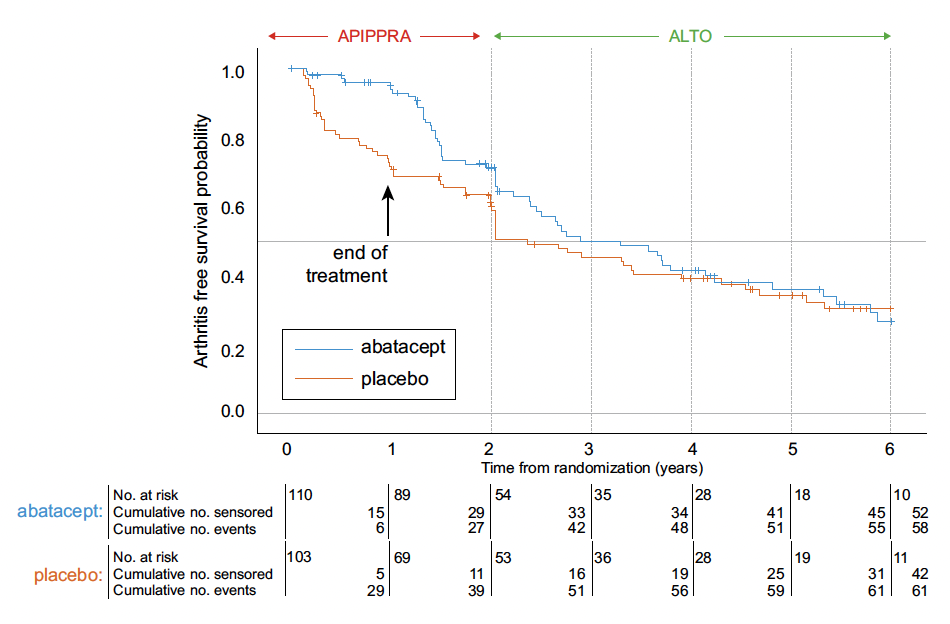
Figure 4 – ALTO : courbe de survie d’une absence de progression vers la polyarthrite rhumatoïde.
Les différences entre les bras en ce qui concerne la durée de survie sans arthrite (définie comme l’aire sous les courbes de survie) étaient de :
• 3,3 mois sur 2 ans (IC 95 % = 1,4-5,3 ; p = 0,001),
• 4,4 mois sur 3 ans (IC 95 % = 1,1-7,8 ; p = 0,008),
• 5,0 mois sur 4 ans (IC 95 % = 0,38-9,8 ; p = 0,039),
• tandis que les différences n’étaient plus maintenues à 5 ans de suivi (5,2 mois, IC 95 % = -1,0-11,4 ; p = 0,102).
L’effet suspensif le plus prolongé de l’ABA sur la survenue d’une PR était observé chez les patients ayant un répertoire auto-immun diversifié, défini par la positivité d’au moins cinq sérotypes d’auto-anticorps différents (ACPA IgG, ACPA IgA, anticorps anti-protéines carbamylées, anticorps anti-protéines acétylées et facteur rhumatoïde) à l’inclusion. Pour ces patients, le bénéfice de l’ABA était maintenu jusqu’à 6 ans (Fig. 5).

Figure 5 – ALTO : probabilité de ne pas avoir d’arthrite en fonction du profil auto-immun diversifié (positivité ou non à au moins cinq sérotypes d’auto-anticorps de la PR) entre les groupes abatacept et placebo.
Tolérance
Cinq événements indésirables graves ont été rapportés dans le groupe ABA contre un dans le groupe placebo au cours du suivi sans qu’aucun ait été considéré comme lié à l’ABA. Sept événements d’intérêt particulier ont été observés dans le groupe placebo et cinq dans le groupe ABA, dont une infection fongique des ongles, considérée comme possiblement liée à l’ABA.
En pratique
L’ABA permet de retarder significativement l’apparition d’une PR chez des patients à haut risque, avec un effet prolongé jusqu’à 6 ans chez ceux présentant un profil auto-immun diversifié sans effet secondaire notable.
• Cope A, Jasenecova M, Vasconcelos J et al. Abatacept in individuals at risk of developing rheumatoid arthritis: results from the arthritis prevention in the pre-clinical phase of RA with abatacept long term outcomes (ALTO) study. Eular 2025 ; OP0004.
TOP 5 – Effets de l’abatacept versus l’hydroxychloroquine sur la progression vers la polyarthrite rhumatoïde chez les patients atteints de rhumatisme palindromique
Les patients atteints de rhumatisme palindromique (RP), une forme d’arthrite/périarthrite intermittente, présentent un risque élevé de développer une PR, en particulier ceux qui sont positifs au FR et/ou aux ACPA. Le traitement du RP est empirique, l’hydroxychloroquine (HCQ) étant l’un des médicaments les plus couramment utilisés en pratique clinique. À ce jour, aucun essai clinique randomisé n’a été réalisé spécifiquement dans le RP. L’abatacept (ABA) pourrait prévenir le développement d’une arthrite persistante ou d’une PR chez les individus présentant des arthralgies inflammatoires ACPA positives.
L’étude PALABA
Dans l’essai PALABA, multicentrique, randomisé, ouvert, mené dans 14 hôpitaux espagnols, les investigateurs ont comparé l’efficacité de l’ABA versus l’HCQ sur la progression vers une PR chez des patients atteints de RP séropositif à début précoce. Les critères d’inclusion étaient : âge ≥ 18 ans, RP selon les critères modifiés de Guerne et Weisman, positivité du FR et/ou des ACPA et une durée des symptômes ≤ 3 ans. Les critères d’exclusion étaient une arthrite persistante dans une ou plusieurs articulations depuis au moins une semaine ou un traitement antérieur par DMARDs.
Les patients étaient randomisés :
• soit dans le bras ABA 125 mg/semaine en sous-cutané durant les 12 premiers mois puis toutes les 2 semaines la deuxième année,
• soit dans le bras HCQ orale (5 mg/kg/jour) avec un suivi de 24 mois.
Le critère principal était la proportion de patients évoluant vers une PR selon les critères de classification ACR/Eular 2010 pendant la période de suivi de 24 mois.
Les patients
Au total, 70 patients (50 femmes, âge moyen 50,9 ± 11,1 ans) ont été inclus dans la population en intention de traiter modifiée : 34 dans le bras ABA et 36 dans le bras HCQ. La durée moyenne des symptômes était de 10 ± 6,6 mois. Le FR et les ACPA étaient positifs chez 63 (90 %) et 57 (81,4 %) patients respectivement (double séropositivité chez 50 patients (71,4 %)).
Résultats
Vingt-six patients du groupe ABA (76,5 %) et 18 patients du groupe HCQ (50 %) ont terminé l’étude. À 24 mois, trois patients (8,8 %) ont développé une PR dans le groupe ABA, contre 10 patients (27,8 %) dans le groupe HCQ (p = 0,042) (Fig. 6). Lors des 12 premiers mois, les proportions de patients ayant évolué vers une PR étaient respectivement de 0 et 22,2 % pour l’ABA et l’HCQ (p = 0,005). La majorité des patients (80 %) traités par HCQ ont progressé vers la PR durant les 12 premiers mois, tandis que, dans le groupe ABA, la progression a été observée après 18 mois de suivi. Les courbes de survie sans PR selon Kaplan-Meier sur 24 mois favorisaient également l’ABA (test log-rank : p = 0,029). La rémission clinique du RP, définie comme l’absence ou un seul épisode palindromique sur une période consécutive de 12 mois après intervention, était plus fréquente chez les patients traités par ABA que ceux sous HCQ (55,9 versus 22,9 % ; p = 0,007).
Les événements indésirables graves étaient rares dans les deux groupes.
En pratique
Chez des patients atteints de RP séropositif à début précoce, l’ABA réduit significativement la progression vers la PR comparé à l’HCQ sur une période de suivi de 24 mois. Cette étude montre que l’ABA est plus efficace que l’HCQ pour suspendre la survenue d’une PR chez les patients présentant un RP, qui est une forme proche d’une pré-PR, confortant les résultats de l’essai APIPRA/ALTO. Cela ne justifie cependant pas l’utilisation de l’ABA en première ligne de traitement dans cette forme précoce de rhumatisme inflammatoire, d’autant que l’utilisation du MTX n’a pas été évaluée pour les patients en échec de l’HCQ !
• Sanmarti R, Perez-Garcia C, de-Toro-Santos FJ et al. Effects of abatacept versus hydroxychloroquine on the progression to rheumatoid arthritis in palindromic rheumatism: a two-year multicenter randomized clinical trial. The PALABA study. Eular 2025 ; OP0105.
Jacques Morel déclare avoir des liens d’intérêt avec Abbvie, Amgen, Biogen, BMS, Boerhinger, Celltrion, Fresenius Kabi, Galapagos, GSK, Janssen, Lilly, Medac, MSD, Novartis, Nordic Pharma, Pfizer, Quiagen, Roche Chugaï, Sandoz, Servier, Theramex, UCB.