Chaque année, depuis 68 ans, les journées de Viggo Petersen permettent de se retrouver et d’échanger sur les pratiques de la rhumatologie et des dernières avancées scientifiques. Cette année, 30 actualités ont été abordées montrant la richesse de notre spécialité et l’interdisciplinarité. Elles s’intéressent aux avancées fondamentales qui ont conduit aux innovations thérapeutiques illustrées par les résultats impressionnants des CAR-T cells dans les maladies auto-immunes réfractaires, aux nouvelles technologies avec l’arrivée submersive de l’intelligence artificielle qui ouvre des perspectives insoupçonnées à la fois diagnostiques, pronostiques et thérapeutiques, et aux pathologies rares et communes.
Comme à chaque édition, les journées de Viggo Petersen sont ponctuées par le jeu concours animé par le Pr Jean-Denis Laredo, que nous remercions de nous instruire avec humour. Mais cette année, le défi a été relevé !
Nous remercions également chaleureusement tous nos collègues qui ont partagé leurs expertises et contribué au succès de ces journées. Un grand merci également aux partenaires de l’industrie qui nous soutiennent depuis toujours. Ce résumé est un aperçu condensé des communications que tous les participants pourront réécouter en replay (bientôt disponible) sur notre site www.viggopetersen.fr.
Nous vous donnons rendez-vous les 20 et 21 mars 2025 pour les 69es journées de Viggo !
Les rhumatismes inflammatoires : de l’alimentation aux cellules CAR-T en passant par le microbiote
Le pré-rhumatisme psoriasique
Le Dr Aline Frazier inaugure les journées sur le concept du pré-rhumatisme psoriasique, en analogie à la pré-polyarthrite rhumatoïde. L’Eular propose plusieurs phases dans l’évolution de l’histoire du rhumatisme psoriasique (RPso) :
• patients à risque de développer un RPso,
• RPso infra-clinique
• et RPso avéré.
Patients à risque de développer un RPso – Le psoriasis atteint 3 % de la population générale, mais seuls 6-11 % développent un RPso. Les atteintes unguéales, des grands plis, du cuir chevelu et cutanées sévères sont associées à un risque plus élevé de développer un RPso ainsi que l’obésité, l’antécédent familial de RPso, certains facteurs génétiques et les uvéites.
RPso infra-clinique – Il concerne les patients atteints de psoriasis avec arthralgie et présence à l’imagerie (échographie, IRM, scintigraphie) de signe inflammatoire des enthèses ou des membranes synoviales, mais sans synovite clinique.
Le concept de pré-RPso a pour objectif principal un diagnostic précoce du RPso et de proposer des traitements permettant de prévenir les lésions ostéoarticulaires. C’est un objectif atteignable dans un futur proche. En effet, la stratégie thérapeutique des collègues dermatologues évolue et vise à réduire les conséquences fonctionnelles du psoriasis avec comme visée thérapeutique un blanchissement cutané, ce qui est maintenant observé avec les nouveaux traitements. Ceux-ci, très efficaces, sur le psoriasis, permettront très probablement de prévenir le développement du RPso.
Les atteintes unguéales du psoriasis et du rhumatisme psoriasique
L’atteinte unguéale du psoriasis peut prendre de nombreux aspects sémiologiques en fonction du site anatomique lésionnel. Le Dr Marine Forien nous rappelle que l’ongle est une véritable interface entre le rhumatologue et le dermatologue, son examen est primordial et révèle parfois des diagnostics de maladies graves telles que connectivites, infections opportunistes et syndrome paranéoplasique.
Atteinte unguéale satellite d’une autre maladie – Ainsi, l’hippocratisme digital doit faire craindre l’ostéoarthropathie hypertrophiante ou syndrome de Pierre Marie et Foix, qui est un syndrome paranéoplasique révélant le plus souvent une pathologie tumorale intra-thoracique. De même, certaines atteintes sont évocatrices de connectivites comme le signe de la manucure au cours de la dermatomyosite ou la présence de méga-capillaires visibles à l’œil nu au niveau du repli sus-unguéal en faveur d’une sclérodermie systémique.
L’atteinte unguéale ou onyxis des RPso, observée jusqu’à 70-80 % des cas, a une sémiologie plus ou moins spécifique.
Atteinte unguéale des RPso – L’onychopachydermopériostose du gros orteil (OP3GO) est une atteinte pathognomonique du RPso. Elle associe une onychopathie psoriasique, un épaississement des parties molles et une périostite de la dernière phalange sans atteinte de l’articulation interphalagienne distale (Fig. 1). Cette atteinte particulière est favorisée par la présence d’un complexe enthésio-synovial et osseux. Ainsi, des structures fibreuses relient le lit de l’ongle au périoste de P3 à proximité de l’insertion du tendon extenseur à la base de P3. Cette atteinte peut être facilement visible par échographie en utilisant une sonde spécifique à haute fréquence. Les atteintes de la matrice du RPso se traduisent par des dépressions ponctuées (« aspect en dé à coudre »), des dépressions transversales, des lignes de Beau, un aspect marbré de la lunule, une trachyonychie et une leuconychie. Les atteintes du lit de l’ongle sont responsables d’onycholyse, de taches saumonées, des hémorragies filiformes en flammèche ou encore d’une hyperkératose sous-unguéale. Le diagnostic différentiel principal est une onychomycose qui peut être favorisée par les traitements immunorégulateurs du rhumatisme inflammatoire et qui représente un cas sur deux des consultations en pathologie unguéale.
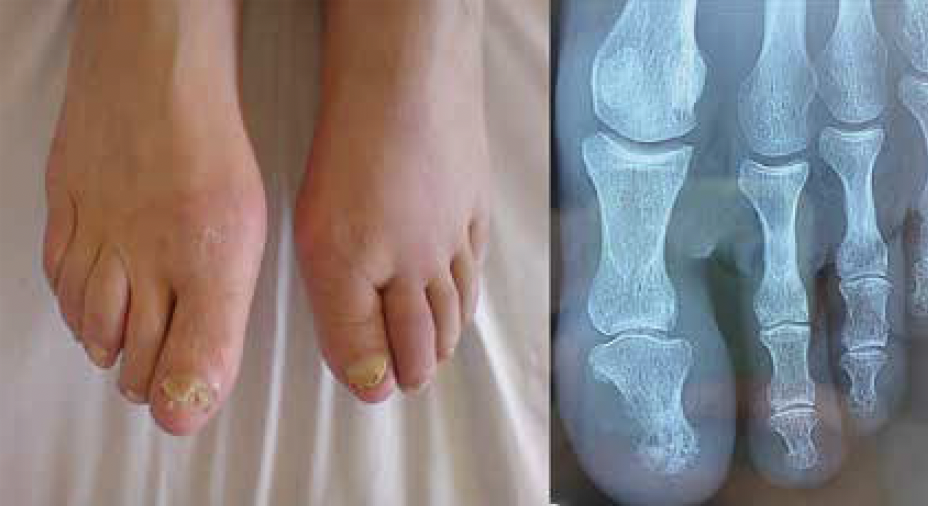
Figure 1 – L’onychopachydermopériostose du gros orteil (OP3GO).
Les traitements par cellules CAR-T
Les traitements par cellules CAR-T (Chimeric antigen receptor) sont une stratégie thérapeutique utilisée depuis plusieurs années en cancérologie avec des résultats parfois impressionnants, permettant une rémission chez 40 % des patients atteints de lymphome diffus à grandes cellules B.
Principe – Ce sont des lymphocytes modifiés génétiquement pour exprimer un récepteur capable de reconnaître un antigène (Ag) spécifique exprimé par les cellules cancéreuses. La modification génétique contient également un élément de co-stimulation qui permet à la cellule CAR-T de s’activer et d’éliminer la cellule cancéreuse une fois liée à elle par la reconnaissance de l’Ag (effet cyto-toxique). Les lymphocytes T des patients sont prélevés puis modifiés génétiquement ex vivo pour exprimer le récepteur spécifique et la co-stimulation. Les cellules CAR-T sont ensuite multipliées pour être ré-injectées chez les patients. Avant cette injection, les patients reçoivent une chimiothérapie pour réduire le système immunitaire afin de diminuer les rejets et permettre aux cellules CAR-T de proliférer. Je vous invite à regarder la présentation du Pr Jérôme Avouac qui fait le point sur l’utilisation de cette stratégie thérapeutique dans les maladies auto-immunes. Une vidéo explique clairement le principe des cellules CAR-T.
Résultats – Cette stratégie a été proposée avec succès chez 15 patients atteints de maladies auto-immunes dépendantes des lymphocytes B, sévères et réfractaires aux traitements disponibles :
• huit lupus érythémateux (LED) avec atteinte rénale glomérulaire type III ou IV,
• trois dermatomyosites (DM)
• et quatre sclérodermies avec atteinte pulmonaire interstitielle (1).
Les cellules CAR-T CD19 injectées ont proliféré rapidement avec un pic observé à 8,6 jours et ont éliminé les cellules lymphocytes B CD19+ au bout de 5,9 jours. Tous les patients ont été améliorés de façon spectaculaire permettant l’arrêt de la corticothérapie et les traitements immunosuppresseurs. Tous les patients lupiques étaient en rémission au 6e mois avec disparition des anticorps anti-DNA et de la protéinurie. La rémission était maintenue malgré la réapparition des lymphocytes B CD19+. Le traitement a été relativement bien toléré, 10 patients ont eu un syndrome de libération cytokinique modéré (grade 1) avec de la fièvre. Le syndrome de libération des cytokines était survenu le lendemain de l’administration des cellules CAR-T CD19 et ont duré en moyenne 5 jours.
Les résultats de cette série de cas suggèrent que la déplétion des lymphocytes B CD19+ au cours des maladies auto-immunes est une piste thérapeutique très prometteuse aboutissant à une rémission soutenue et persistante. Une reconstitution complète des lymphocytes B était observée, sans rechute de la maladie auto-immune, montrant donc une modification des phénotypes des lymphocytes B après le traitement, en quelque sorte « une réinitialisation du système immunitaire ».
À suivre – Cette stratégie thérapeutique est en pleine expansion et d’autres constructions sont testées comme les CAR-Tregs (T régulateurs modifiés avec un récepteur chimérique). Ainsi, dans la polyarthrite rhumatoïde, l’induction de la tolérance dans la membrane synoviale des articulations atteintes par le développement de CAR-Tregs spécifiques de protéine citrullinée donne des résultats prometteurs.
Qu’en est-il de l’immunité innée ?
La modulation de l’immunité humorale permet donc de lutter contre les cancers et les maladies auto-immunes. Peut-on utiliser le même raisonnement pour l’immunité innée ?
Immunité innée et mémoire immunitaire – La mémoire innée constitue la première ligne de défense immunitaire et dépend des monocytes, des macrophages, des neutrophiles, des cellules dendritiques, des cellules NK et des cellules lymphoïdes innées. Des observations anciennes suggèrent que l’immunité innée possède aussi une mémoire immunitaire comme l’immunité adaptive. Une meilleure réponse immunitaire au cours d’une réinfection, correspondant de facto à une mémoire immunitaire, est décrite depuis longtemps chez les invertébrés et les plantes.
L’immunité entraînée – Dans ma communication, je mets en avant des travaux récents qui montrent que l’immunité innée peut être entraînée pour lutter contre les infections ou les cancers. À l’inverse, elle peut être inhibée (induction d’une tolérance) au cours des maladies auto-inflammatoires ou auto-immunes. Le concept de l’immunité entraînée (trained immunity) correspond aux adaptations à long terme des cellules de l’immunité innée induites par des stimuli endogènes et/ou exogènes, à l’origine d’une réponse immunitaire cellulaire renforcée lors d’une seconde stimulation par le même pathogène ou un autre (2).
Les adaptations cellulaires – Ces adaptations cellulaires comprennent des modifications épigénétiques, transcriptomiques et métaboliques. Elles touchent les cellules immunitaires périphériques et centrales, i.e. les monocytes et neutrophiles circulants, les macrophages tissulaires et les précurseurs hématopoïétiques de la moelle osseuse. Ces modifications se produisent dans les précurseurs des cellules immunitaires de la moelle osseuse et sont transmises aux cellules immunitaires circulantes. Elles se produisent aussi dans les macrophages résidents tissulaires. De nombreux motifs microbiens et de lésions peuvent moduler de façon plus ou moins prolongée le phénotype pro-inflammatoire ou tolérant des cellules de l’immunité innée. Par exemple, la vaccination par le BCG induit des modifications épigénétiques des précurseurs hématopoïétiques qui persistent pendant plus de 1 an. Cette adaptation cellulaire varie en fonction de la durée et de la quantité du stimulus.
Les modifications épigénétiques – Les modifications épigénétiques comprennent les méthylations/déméthylations des cytosines de l’ADN, des méthylations et acétylations des résidus lysine des histones qui compactent l’ADN. Elles modifient l’accessibilité des promoteurs des gènes et par conséquent leur transcription. Ainsi, la mono- ou tri-méthylation de la lysine 4 de l’histone 3 (H3K4me1, H3K4me3) ou l’acétylation de la H3K27 (H3K27ac) des promoteurs des gènes inflammatoires IL-1b, IL-6 et TNF-a des monocytes/macrophages immuno-entraînés augmentent la production de ces cytokines inflammatoires. Inversement, les modifications épi-
génétiques H3K9me3 ou H3K9me2 sur les promoteurs de ces mêmes gènes répriment leur expression. Les ARN longs non codants (lncRNA) qui régulent la transcription des gènes sont aussi impliqués dans la régulation épigénétique des cellules immunitaires innées en modulant H3K4me3. Ces modifications épigénétiques sont associées et modulées par des modifications des métabolismes cellulaires. L’accumulation des métabolites du cycle de Krebs comme le citrate, le succinate et le fumarate participent aux modifications épigénétiques.
L’immunité cellulaire entraînée/expérimentée est bénéfique dans la défense anti-microbienne. Une activation accrue peut contribuer à une inflammation chronique comme observée au cours de la polyarthrite rhumatoïde, des rejets de greffe, des maladies allergiques, auto-immunes et auto-inflammatoires. De même, une réponse immunitaire cellulaire atténuée, i.e. une immuno-tolérance, peut favoriser le développement de cellules infectées et/ou cancéreuses. Le développement de traitements capables de moduler les modifications métaboliques et épigénétiques de l’immunité innée constitue un grand champ d’application dans ces maladies (2).
Microbiote respiratoire et polyarthrite rhumatoïde
La citrullination des protéines et la production des anticorps anti-peptides citrullinées (ACPA) peuvent être modulées par des facteurs génétiques et environnementaux tels que le tabac et le microbiote. Le rôle du microbiote pulmonaire dans la polyarthrite rhumatoïde (PR) prend de l’ampleur grâce aux progrès technologiques dans l’identification des espèces et la possibilité de l’étudier sur des prélèvements non invasifs (expectoration induite).
Le microbiote pulmonaire – Le Dr Pierre-Antoine Juge nous rappelle que le microbiote respiratoire est moins dense et moins riche que celui du tube digestif et de la peau, constitué par près de 50 % de bactéries anaérobies (anaérobiome), et notamment des genres Prevotella, Veillonella, Fusobacterium ou Porphyromonas. Le microbiote respiratoire est dynamique, résultant des microbes environnementaux et digestifs, de la réponse immunitaire et de la clairance mucociliaire.
La dysbiose respiratoire – Une réduction de la diversité bactérienne traduit une dysbiose respiratoire et est observée dans plusieurs maladies pulmonaires telles que la broncho-pneumopathie chronique obstructive, la mucoviscidose ou la fibrose pulmonaire idiopathique. Il est difficile de savoir si cette dysbiose est la cause ou la conséquence de ces pathologies. Les modifications architecturales pulmonaires et les défauts de clairance mucociliaires observés au cours de ces pathologies peuvent être à l’origine d’une perturbation du microbiote respiratoire. À l’inverse, la dysbiose peut activer certains signaux inflammatoires (voies NF-kB, de l’IL-17 ou des PI3 kinases) de la PR.
Dysbiose respiratoire et PR – Au cours des PR récentes, on observe une diminution de plus de 40 % de la diversité microbienne avec la quasi-disparition de familles bactériennes telles que les Actinomycetaceae et les Spirochaetaceae. L’enrichissement de certaines souches bactériennes, notamment Haemophilus et Streptococcus, est corrélé à la présence des ACPA.
Cependant, ces résultats nécessitent confirmation. Les études actuelles n’évaluent pas l’effet des facteurs pouvant modifier le microbiote comme le tabac et les traitements immuno-modulateurs. Il reste donc un long chemin avant de pouvoir comprendre les relations exactes de cause à effet entre dysbiose respiratoire et PR.
Rôle de l’alimentation
Si les modulations du microbiote respiratoire pour contrôler l’activité de la PR restent difficiles, qu’en est-il de l’alimentation ?
Les recommandations françaises – Le Pr Jérémie Sellam, qui a coordonné les recommandations françaises sur l’alimentation des patients souffrant de rhumatisme inflammatoire, rappelle quelques points importants :
• les modifications de l’alimentation ne peuvent pas se substituer au traitement de fond ;
• insister sur la perte de poids chez des patients obèses ou en surpoids, et expliquer ses effets bénéfiques articulaires, métaboliques, cardiovasculaires (CV) et psychiques ;
• éviter les régimes d’exclusion chez des patients non allergiques. Il n’existe aucune preuve sur les effets bénéfiques de ces régimes qui peuvent, à l’inverse, être responsables de carence nutritionnelle, dont la vitamine B12 ;
• favoriser une alimentation équilibrée et diversifiée, pauvre en protéine animale, type régime méditerranéen, qui est riche en produits céréaliers complets, légumes, fruits, huile d’olive, légumineuses, et limité dans les apports en viande et graisse d’origine animale.
L’alimentation est un sujet important à discuter avec les patients, car plus de 26 % des patients sondés déclarent suivre ou avoir suivi un régime d’exclusion et 42 % pensent que l’alimentation agit sur les symptômes et les douleurs articulaires. Il est nécessaire de leur fournir des informations justes.
Méthotrexate : les bonnes pratiques
Les effets de l’alimentation sur les rhumatismes inflammatoires nécessitent encore des études bien conduites. En revanche, ceux du méthotrexate (MTX) sont bien connus. Le Pr Frédéric Lioté fait une mise au point sur les bonnes pratiques de sa prescription.
Méthotrexate et microbiote intestinal – Il rappelle que le MTX pourrait aussi avoir des effets sur le microbiote intestinal, en favorisant des bactéries productrices de folates qui peuvent moduler l’inflammation intestinale.
Contre-indications – Le MTX est contre-indiqué pendant la grossesse et chez des patients avec maladie rénale sévère stade 4/5, définie par une clairance de la créatinine < 30 ml/min/1,73 m2.
Avant l’initiation – Il est conseillé de mettre à jour les vaccinations (pneumocoque, DTP, grippe et SARS CoV-2) et de faire une « recharge » en folates pour améliorer la tolérance du traitement.
L’acide folique – La co-prescription MTX/acide folique améliore de plus de 70 % la tolérance du MTX. Elle diminue de 70-85 % les effets secondaires gastro-intestinaux et de 29-35 % les effets secondaires hépatiques. La dose d’acide folique est de 5-10 mg/semaine, quelle que soit la dose du MTX. L’acide folique peut être administré à distance du MTX, généralement 48 heures avant ou après.
Posologie – Le MTX est initié à 10 ou 15 mg/semaine et augmenté de 2,5 ou 5 mg tous les 2 mois jusqu’à obtention de la rémission du rhumatisme inflammatoire. La dose maximale autorisée est de 25 à 30 mg/semaine. Au-delà de 15 mg/semaine, il est recommandé d’administrer le MTX en deux prises le même jour, pour augmenter la biodisponibilité (d’environ 25 %). L’autre moyen pour augmenter sa biodisponibilité est l’administration en injection sous-cutanée.
Tolérance – En général, le MTX est bien toléré. Les données récentes montrent que, non seulement, il n’augmente pas le risque de fibrose pulmonaire, mais, au contraire, il ralentit et réduit la progression d’une pneumopathie interstitielle. Les effets secondaires principaux sont gastro-intestinaux, hématologiques et hépatiques qu’il convient de surveiller par des analyses sanguines. Devant une cytolyse hépatique, il convient d’éliminer les autres causes fréquentes que sont les toxiques (alcools, autres médicaments), le syndrome métabolique (MASH ou Metabolic-Associated SteatoHepatitis) et les infections virales.
Les comorbidités dans la polyarthrite rhumatoïde
La PR s’accompagne de nombreuses comorbidités qu’il convient de dépister régulièrement, en particulier les maladies CV, l’ostéoporose, certains cancers, les infections et les troubles thymiques.
Risque de mortalité augmenté – Ces comorbidités, l’inflammation systémique de la PR et les traitements augmentent le risque de mortalité des patients atteints de PR comme nous le rappelle le Dr Anna Molto. Les études sur la base nationale des données de santé (SNDS) en France et celle des vétérans aux États-Unis montrent un risque de mortalité globale augmenté de 29 % (taux standardisé de mortalité par rapport à la population générale de 1,29 (IC 95 % 1,28-1,31)) et de 23 % (IC 95 % 1,20-1,26), respectivement, en particulier pour les mortalités liées aux maladies CV, pulmonaires et aux infections.
Le dépistage, la prévention et la prise en charge des comorbidités sont indispensables pour diminuer ce risque et doivent être effectués annuellement soit au cours d’une consultation spécifique, soit au cours d’une hospitalisation. Le rhumatologue doit coordonner cette prise en charge. Il peut s’aider d’une infirmière spécialisée ou de coordination. Et de façon plus efficace, il doit éduquer et impliquer le patient pour qu’il devienne actif dans cette prise en charge.
La pneumopathie interstitielle diffuse
Le poumon est l’atteinte extra-articulaire la plus fréquente de la PR. La présence d’une pneumopathie interstitielle diffuse (PID) est associée à une augmentation de la mortalité (HR 3,4 ; IC 95 % 3,1-3,9) proche de celle de la fibrose pulmonaire idiopathique. Peut-on la dépister avec l’échographie pulmonaire ?
L’échographie pulmonaire – Revoyez l’exposé du Dr Sébastien Ottaviani qui nous montre les différentes images à retenir, dont les lignes A et Z qui sont des artéfacts de réverbération de la ligne pleurale et de l’épaississement focal des septa interlobulaires, respectivement. L’échographie pulmonaire pourra détecter et quantifier un épanchement pleural et aussi guider son aspiration, un épaississement de la ligne pleurale et la présence des lignes B.
Ces lignes sont évocatrices d’une PID, elles correspondent à des artéfacts de réverbération de la ligne pleurale due à un épaississement des septa interlobulaires. Elles se visualisent sous forme d’images hyperéchogènes perpendiculaires à la ligne pleurale, donnant un aspect en queue de comète ou projecteur et effaçant la ligne A (Fig. 2). Leur nombre est proportionnel à l’importance de l’atteinte interstitielle. Les signes échographiques sont bien corrélés au scanner thoracique. La sensibilité et la spécificité de l’échographie pour détecter une PID sont de 92 et 89 % (3).
L’échographie apparaît donc comme un outil simple et non irradiant pour détecter les atteintes interstitielles pulmonaires et les épanchements pleuraux. Examen qui ne substitue pas au scanner qui reste l’examen de référence.
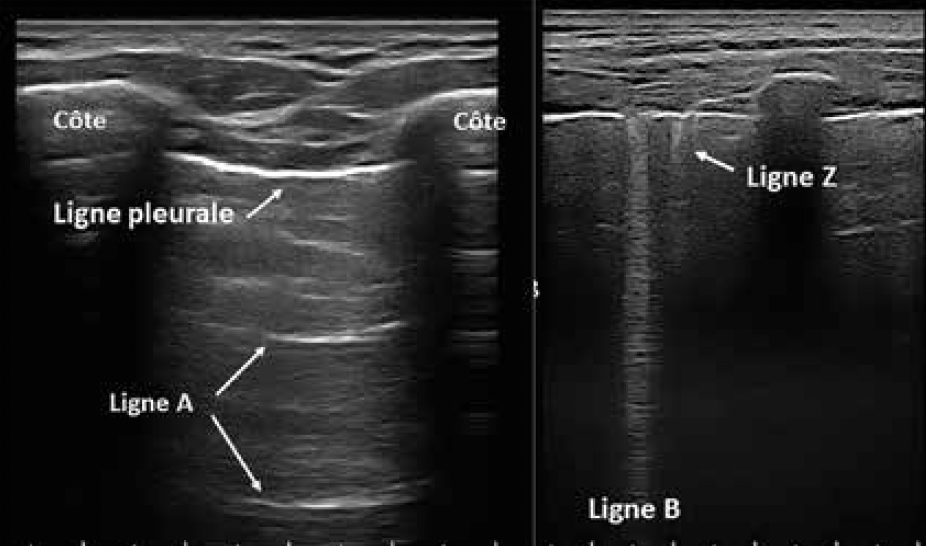
Figure 2 – Les signes échographiques normaux (lignes A et Z) et évocateurs de pneumopathie interstitielle (ligne B). Notez l’aspect en queue de comète ou de projecteur de la ligne B.
Les pathologies microcristallines
Le risque cardiovasculaire
L’augmentation de la mortalité CV est aussi observée dans les pathologies microcristallines comme le soulignent les Prs Tristan Pascart et Thomas Bardin.
Dans la goutte – Elle est favorisée par les nombreuses comorbidités et l’inflammation liée aux dépôts des cristaux d’urate de sodium, en particulier lors des crises inflammatoires. Deux études récentes montrent que les crises de goutte sont associées à un risque augmenté de plus de 70 % d’infarctus du myocarde et d’accidents vasculaires cérébraux avec un risque accru qui persiste pendant 4 mois (4). De même, les crises inflammatoires liées aux cristaux de pyrophosphate sont associées à un risque augmenté d’événement CV.
Physiopathologie – Ces crises inflammatoires microcristallines produisent de nombreuses cytokines inflammatoires, dont l’IL-1β, qui pourraient déstabiliser les plaques d’athérome en activant l’infiltrat inflammatoire, la production de métalloprotéases, l’ulcération des plaques et la formation du thrombus. Celle-ci est aussi favorisée par l’hypercoagulabilité liée à l’inflammation aiguë et la formation des TRAP par les neutrophiles.
La colchicine – Le rôle de l’inflammation dans les événements CV est fortement suggéré par les essais cliniques CANTOS et LODOCO montrant qu’un traitement prolongé par canakinumab ou colchicine réduit les événements CV chez des patients à haut risque, y compris les patients non goutteux (5-7). Ces études ont conduit la FDA à approuver l’utilisation de la colchicine dans cette indication. Les sociétés savantes de cardiologie discutent dans leurs recommandations l’ajout de la colchicine chez des patients avec lourds antécédents CV.
Le fébuxostat – Le risque CV du fébuxostat, suggéré par l’étude CARES chez des patients américains à très haut risque, n’est ni confirmé par l’étude européenne FAST, qui est méthodologiquement meilleure (moins de perdus de vue), ni par les récentes méta-analyses.
Les traitements hypouricémiants – Les risques CV semblent plus liés aux crises inflammatoires qu’il convient de prévenir en initiant les traitements hypouricémiants (THU) à faible dose associée à une prophylaxie prolongée par colchicine, pendant au moins 6 mois. Ce risque CV devrait diminuer et disparaître sous THU bien conduit. En effet, les crises inflammatoires disparaissent après la dissolution des cristaux d’urate.
Comme pour la PR, la prise en charge et le dépistage des comorbidités sont indispensables pour diminuer ce risque, et devraient aussi impliquer des infirmières et les patients. Le traitement de ces comorbidités doit favoriser l’utilisation de médicaments doués de propriété hypouricémiante comme le losartan, les inhibiteurs calciques, le fénofibrate et les inhibiteurs des co-transporteurs sodium/glucose de type 2 (SGLT2i).
Les SGLT2i – Les SGLT2i diminuent de façon modeste l’uricémie (environ 50 µmol/l) et ont des propriétés anti-inflammatoires (8, 9). Ils diminuent la goutte incidente chez des patients diabétiques et le nombre de crises chez des patients goutteux diabétiques. Ces traitements diminuent la mortalité globale et CV. Ils sont indiqués pour le diabète de type 2, l’insuffisance cardiaque et la maladie rénale chronique qui sont des comorbidités fréquentes des patients goutteux.
Les traitements hypouricémiants
Le THU doit être augmenté progressivement jusqu’à l’obtention de l’uricémie cible inférieure à 300 µmol/l (50 mg/l), ce qui permet la dissolution des cristaux d’urate et par conséquent la guérison de la maladie et très probablement la diminution des risques CV.
L’allopurinol – Il reste le THU de première intention selon les recommandations nationales et internationales chez des patients avec une clairance de la créatinine > 45 ml/min/1,73 m2. Il est débuté à faible dose (100 mg/jour chez un patient avec une fonction rénale normale, et 50 mg/jour chez un patient avec une clairance < 60 ml/min/1,73 m2), à distance d’une crise, avec une prophylaxie prolongée d’au moins 6 mois. Chez des patients avec une clairance < 45 ml/min/1,73 m2, le fébuxostat est indiqué et initié aussi à faible dose (40 mg/jour).
L’allergie cutanée – Le principal risque de l’allopurinol est l’allergie cutanée grave qui survient dans les 3 premiers mois du traitement. Les facteurs de risque liés à cette allergie sont la présence de l’antigène HLAB5801, une dose initiale d’allopurinol trop élevée par rapport à la clairance de la créatinine, l’utilisation de diurétique et la maladie rénale stades 4/5. La présence de l’antigène HLAB5801 doit faire éviter l’utilisation de l’allopurinol.
Intolérance – En cas d’intolérance aux deux inhibiteurs de la xanthine oxydase, le probénécide est un uricosurique qui est à nouveau disponible en pharmacie de ville. La benzbromarone (Narcaricin®) est uniquement disponible en pharmacie hospitalière après obtention de l’autorisation d’accès compassionnel (AAC).
D’autres uricosuriques et uricases sont en développement ou déjà disponibles au Japon et aux États-Unis.
La maladie à dépôt de cristaux de pyrophosphate de calcium
Si la goutte est curable, la maladie à dépôt de cristaux de pyrophosphate de calcium (PPC), encore appelée chondrocalcinose, reste une maladie sans traitement efficace pour dissoudre ces cristaux. Plusieurs actualités marquantes ont été réalisées en 2023, avec en particulier les résultats de l’essai clinique COLCHICORT, mené par le Pr Tristan Pascart, dans la crise de chondrocalcinose (10).
Colchicine versus prednisone – La colchicine à faible dose (1,5 mg le premier jour) est aussi efficace que la prednisone 30 mg/jour après 24 heures de traitement pour les crises de chondrocalcinose évoluant depuis moins de 36 heures. Dans cette étude multicentrique randomisée, environ deux tiers des 109 patients traités étaient améliorés, mais 22 % des patients sous colchicine avaient eu de la diarrhée versus 6 % des patients sous corticoïde. Inversement, une plus grande proportion de patients sous corticoïde ont eu des troubles psychiques (excitation 4 %, euphorie 1 %, insomnie 4 %), un déséquilibre glycémique (6 %) ou une poussée hypertensive (11 %) par rapport aux patients sous colchicine (26 versus 2 % pour tous ces effets secondaires).
Cette première étude montre que l’efficacité des deux médicaments est très rapide et qu’il est donc judicieux de réduire leur durée de prescription pour améliorer la tolérance thérapeutique.
L’anakinra – Des études de cas de série suggèrent l’efficacité de l’anakinra, le récepteur antagoniste recombinant de l’IL-1, qui peut être proposé en cas d’intolérance ou de contre-indication à la colchicine, aux AINS et aux corticoïdes.
Dans les formes inflammatoires chroniques – On ne dispose pas de données solides. Le méthotrexate n’a pas montré d’efficacité dans une étude randomisée en cross-over. L’inhibition de l’IL-6 par le tocilizumab semble être une piste prometteuse, suggérée par une étude pilote menée par le Dr Augustin Latourte, dans laquelle il a observé une amélioration de plus de 80 % chez 11 patients qui étaient réfractaires aux autres traitements (11). Un PHRC coordonné par l’équipe de Lariboisière va tester cette hypothèse en évaluant l’efficacité de ce traitement dans les formes chroniques inflammatoires.
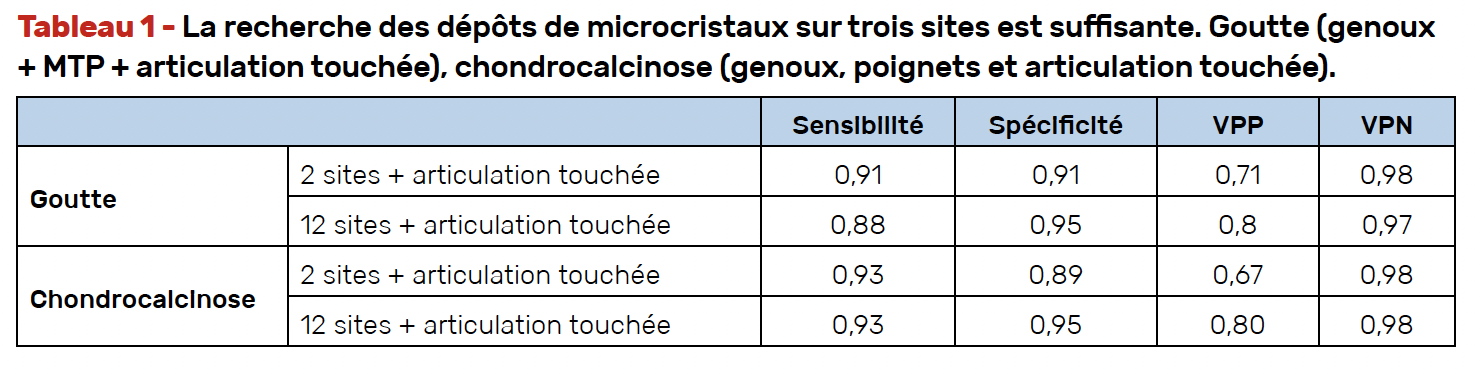
Diagnostic et classification – Les autres actualités marquantes sont l’élaboration des critères diagnostiques et de classification ACR/Eular et les recommandations sur l’imagerie dans le diagnostic des dépôts de PPC. L’échographie est l’examen de choix avec une meilleure sensibilité par rapport à la radiographie standard. Aux poignets, sensibilité et spécificité de l’échographie sont respectivement de 87 et 87 % versus 67 et 95 % pour la radiographie. Aux genoux, elles sont de 85 et 91 %. La recherche des dépôts cristallins (PPC et urate) sur des sites cibles est aussi performante que lorsqu’elle est effectuée de façon extensive sur plusieurs sites. La sensibilité et spécificité de la recherche sur deux articulations (genoux et métatarsophalangiennes du premier rayon pour la goutte, genoux et poignets pour la chondrocalcinose) et l’articulation symptomatique sont, respectivement, pour la goutte de 91 et 91 % et pour la chondrocalcinose de 93 et 89 % (Tab. 1) (12, 13).
Les formes familiales – Autre point d’actualité, le Pr Pascal Richette revisite les formes familiales des maladies à dépôts de pyrophosphate de calcium. La survenue d’une chondrocalcinose avant 60 ans doit faire rechercher une hyperparathyroïdie, une hémochromatose, une hypomagnésémie, une hypophosphatasie et une forme familiale. Deux gènes sont associés à la chondrocalcinose familiale : ANKH et TNFSF11B codant, respectivement, pour les protéines ANKH et ostéoprotégérine. Les mutations perte de fonction du TNFSF11B ont été décrites dans trois familles et sont responsables de polyarthropathies, arthrose-like, avec dépôts de PPC. Les mutations gain de fonction du gène ANKH ont été décrites dans plusieurs familles et sont associées à des formes inflammatoires et des arthropathies ankylosantes avec dépôts de PPC. Les données récentes montrent que la protéine ANKH transporte l’ATP et le citrate. Son activité de transport du pyrophosphate est remise en question. Le métabolisme de l’ATP exporté vers le milieu extra-cellulaire est responsable d’une accumulation du pyrophosphate extra-cellulaire à l’origine de la formation des cristaux. Les mécanismes de formation des cristaux associés aux mutations du TNFSF11B ne sont pas connus.
L’arthrose
Le lien exact entre arthrose et cristaux de PPC n’est pas élucidé. C’est la question de la poule ou de l’œuf… i.e. est-ce la destruction du cartilage au cours de l’arthrose qui favorise la formation de ces cristaux ou inversement la présence de ces cristaux qui accélère la destruction du cartilage ?
Des données cliniques et fondamentales sont en faveur d’un rôle arthrogène des cristaux de PPC, en particulier la survenue d’arthrose des articulations habituellement épargnées par l’arthrose primitive telles que la cheville, le poignet, l’épaule et le coude dans les formes secondaires ou sévères de dépôts de cristaux de PPC.
L’arthrose du coude
En effet, une atteinte arthrosique de ces différentes articulations est rarement primitive comme le rappelle le Dr Leslie Benattar pour le coude.
Épidémiologie – L’arthrose du coude représente 1 à 2 % de toutes les pathologies du coude. Elle prédomine chez l’homme, avec une moyenne d’âge au diagnostic de 50 ans, et touche le bras dominant dans 80-90 % des cas. Le compartiment huméro-radial est le premier atteint, en raison de contraintes mécaniques plus importantes.
L’arthrose primitive – Elle est rare, liée à une sursollicitation d’origine sportive ou professionnelle avec microtraumatismes répétés et/ou port de charges lourdes. On la retrouve en particulier dans les sports de lancer, de soulever de poids, et d’arts martiaux.
La forme secondaire – Celle-ci est la plus fréquente. Elle peut être due à une infection, une maladie auto-immune, une maladie métabolique de surcharge (hémochromatose, maladie de Wilson, ochronose), une ostéochondrite primitive du capitellum (ou maladie de Panner), une ostéochondrite disséquante du capitellum ou encore à des dépôts de PPC.
L’ostéochondrite primitive de Panner – Il s’agit d’une maladie de croissance qui touche l’enfant de 6 à 10 ans, volontiers sportif avec existence de sports à risque. Elle est due à la fragmentation du noyau d’ossification du capitellum avec nécrose de l’os sous-chondral. Elle se manifeste par un déficit d’extension, une douleur latérale sans blocage. La prise en charge est médicale avec arrêt de l’activité sportive jusqu’à la disparition des douleurs.
Prise en charge de l’arthrose du coude – Elle est identique aux autres articulations en privilégiant un traitement conservateur. Celui-ci associe des mesures pharmaceutiques et non pharmaceutiques, de la rééducation, une adaptation des activités sportives et un dépistage d’une compression du nerf ulnaire.
Au cours de l’arthrose, existe-t-il des pistes thérapeutiques pour régénérer le cartilage détruit ?
Question à laquelle tente de répondre le Dr Augustin Latourte.
Les gènes associés à l’arthrose – Les études pan-génomiques (GWAS) récentes ont identifié des polymorphismes de 80 gènes associés à l’arthrose. Ces gènes codent pour des protéines de la matrice du cartilage, des facteurs de croissance et des protéines qui régulent le développement suggérant donc un important rôle des facteurs qui pourraient réguler la réparation du cartilage.
Un défaut de réparation du cartilage – L’arthrose serait ainsi secondaire à un défaut de réparation du cartilage, et pas uniquement à un excès de dégradation du tissu. Cette hypothèse est étayée par l’augmentation de l’interligne articulaire des genoux arthrosiques, et donc de l’épaisseur du cartilage, après 6 semaines de distraction articulaire chirurgicale.
La sprifermine – D’autres pistes moins agressives apportent des résultats prometteurs. Ainsi, la sprifermine, une protéine recombinante du FGF18, stoppe la perte du cartilage fémoro-tibial, voire augmente son épaisseur après quatre cycles de traitement sur 18 mois.
Les cellules souches – L’autre possibilité pour stimuler la réparation du cartilage est le recrutement des cellules souches. Une étude récente a identifié l’angiopoïetine-like 3 (ANGPTL3) comme facteur capable d’induire une différenciation chondrocytaire (14). L’extrémité C-terminale recombinante de cette molécule, appelée LNA043, induit la différenciation chondrogénique des cellules souches, et stimule la production de matrice cartilagineuse. Cette molécule est capable de diminuer les lésions cartilagineuses dans un modèle d’arthrose induite chez le rat et de réparer les lésions cartilagineuses dans un modèle de lésion focale chez le cochon nain. Chez l’homme, l’analyse des pièces opératoires de patients opérés d’une prothèse totale de genou et qui avaient eu 21 jours avant la chirurgie des injections intra-articulaires de LNA043 a montré que i) le traitement a effectivement pénétré le cartilage articulaire et ii) qu’il a modifié le profil transcriptomique des chondrocytes. Les chondrocytes traités par LNA043 ont augmenté l’expression de gènes codant pour les constituants de la matrice extra-cellulaire du cartilage et de voies de signalisation pro-anaboliques, tout en diminuant celle de gènes codant pour des médiateurs délétères dans la physiopathologie de l’arthrose.
Le LNA043 est donc un candidat de choix pour un traitement pro-régénératif dans l’arthrose, puisque cette molécule est non seulement capable d’avoir un effet sur le métabolisme des chondrocytes vers un profil pro-anabolique, mais aussi de promouvoir la différenciation chondrogénique des cellules souches résidentes articulaires. Ces résultats encourageants ont conduit les auteurs à mettre en place une étude de phase IIb. Ces traitements anaboliques pourront-ils améliorer la douleur et la fonction des patients ? Ces objectifs thérapeutiques représentent un véritable défi pour la prise en charge de l’arthrose.
L’embolisation des artères géniculées
Une nouvelle piste, exposée par le Dr Thomas Léger, donne des résultats prometteurs : l’embolisation des artères géniculées. Ce traitement mini-invasif est fondé sur l’hypothèse d’un rôle de la néo-angiogenèse, présente au cours de l’arthrose, dans la genèse des douleurs de l’arthrose. Les premières embolisations d’artères géniculées ont été décrites dans le cadre d’hémarthroses récidivantes.
Dans la gonarthrose – En 2015 est publiée la première étude évaluant cette technique chez 11 patients souffrant de gonarthrose et traités entre 2012 et 2013 avec des résultats encourageants à 4 mois. Tous les patients présentaient des néovaisseaux en angiographie, sous forme de « blush vasculaire », de siège généralement corrélé au territoire douloureux et en rapport avec la synovite.
Dans la gonarthrose stade KL 1-3 – Par la suite, la même équipe a rapporté son expérience chez 72 patients souffrant de gonarthrose stade Kellgren-Lawrence (KL) 1-3, avec de bons résultats cliniques, objectivés par une baisse significative et prolongée du score WOMAC avec plus de 2 ans de suivi. Les taux de succès cumulés à 6 mois et 3 ans étaient de 86,3 % (15). Les IRM de 35 genoux à 2 ans ont montré une amélioration significative de la synovite par rapport à l’état initial.
La technique nécessite une bonne connaissance de l’anatomie mais, dans les mains expertes, le taux de succès est élevé du fait de l’anatomie des artères géniculées globalement favorable au micro-cathétérisme. Revoyez la présentation, les images d’embolisation des artères sont magnifiques.
Le matériel – Le matériel d’embolisation n’est pas consensuel. Il s’agit soit de l’imipénème cilastatine (cristaux résorbables de 10-70 μm), soit de microsphères non résorbables (généralement d’un diamètre allant de 75 à 300 μm).
Les complications – Les complications sont peu fréquentes et de faible gravité, principalement cutanées (changement de couleur transitoire, très rarement des ulcérations), ou hématome du point de ponction.
L’embolisation des artères géniculées dans la gonarthrose semble donc être une piste intéressante et à faible risque. Une étude randomisée multicentrique, coordonnée par le Pr Christian Roux, CHU de Nice, et à laquelle l’équipe de Lariboisière participe, apportera une réponse définitive.
L’ostéoporose
La prise en charge de l’ostéoporose est défaillante en France, due à un défaut de dépistage, de traitement qui est freiné par une diffusion large des effets secondaires graves potentiels, pourtant peu fréquents. Il paraît ainsi important de communiquer sur les effets bénéfiques à la fois osseux et extra-osseux.
Les conséquences socio-économiques
Dr Arnaud Vanjak rappelle que l’ostéoporose, maladie caractérisée par une masse osseuse basse avec altérations micro-architecturales exposant au risque de fracture, a des conséquences socio-économiques graves. L’ostéoporose touchait environ 4 000 000 patients en France en 2019, et est responsable de presque 500 000 fractures incidentes, ce qui représente environ 7 milliards de coûts directs.
Les traitements ostéoformateurs et anti-résorptifs
Les traitements de l’ostéoporose ostéoformateurs (tériparatide, romosozumab) ou anti-résorptifs (bisphosphonates, dénosumab) ont une efficacité anti-fracturaire confirmée par de nombreux essais cliniques et méta-analyses.
Les bisphosphonates – (Zolédronate dans les essais randomisés, risédronate et alendronate dans les études observationnelles), en plus de leur effet antifracturaire, ils diminuent la mortalité de 25 à 35 % selon des études (16, 17). Cette diminution de la mortalité est expliquée par l’effet antifracturaire, mais probablement aussi par une diminution de la mortalité CV et de l’incidence des cancers. Par exemple, une étude montre que l’utilisation de bisphosphonates chez des patientes ménopausées avec un diagnostic précoce de cancer du sein diminue la mortalité liée à la maladie (HR 0,82 ; IC 95 % 0,73-0,93) ainsi que le risque de récurrence (HR 0,86 ; IC 95 % 0,78-0,94). Les mécanismes impliqués ne sont pas encore bien compris. Les données précliniques suggèrent une action cellulaire sur les monocytes/macrophages ainsi qu’un effet direct et indirect sur les cellules musculaires lisses vasculaires, les cellules endothéliales et les cellules tumorales.
Le raloxifène – Le traitement par raloxifène chez des femmes ménopausées de moins de 80 ans est aussi associé à une diminution de l’incidence des cancers du sein avec un risque relatif de 0,24 (IC 95 % 0,13-0,44).
Le dénosumab – Enfin, le dénosumab, anticorps monoclonal anti-RANKL, ne semble pas avoir d’effet sur la mortalité. En revanche, une étude cas-témoin montre que le traitement par dénosumab est associé à une incidence diminuée du diabète de type 2 par rapport à un traitement par bisphosphonate oral. Il augmente aussi la force musculaire.
Il n’y a pas actuellement de données sur les effets extra-osseux des traitements ostéoformateurs. Ces données bénéfiques extra-osseuses doivent être communiquées aux patients pour augmenter leur adhésion thérapeutique.
Est-ce que les traitements à visée osseuse altèrent la consolidation et la croissance osseuse ?
Question abordée par le Pr Martine Cohen-Solal qui rappelle que cette préoccupation existe depuis l’arrivée de ces traitements. Quel que soit l’âge, le maintien d’une activité de modelage chez l’enfant et du remodelage chez l’adulte est indispensable à assurer la croissance et la réparation osseuse. L’inhibition prolongée du remodelage osseux dans les deux situations a fait évoquer un effet négatif potentiel et a suscité quelques études sur l’utilisation des traitements anaboliques. Les études sur ces questions apportent des réponses rassurantes.
Chez l’enfant – Les données sont issues des patients avec ostéo-genèse imparfaite. Chez 552 adolescents d’âge moyen de 12 ans avec ostéogenèse imparfaite de type 1 à VI, la taille moyenne est inférieure au 3° percentile et reste en dessous à l’âge adulte (18). Cependant, la courbe de croissance et la taille finale n’étaient pas corrélées à la prise de bisphosphonates.
Chez l’adulte – Plusieurs études et une méta-analyse sur série de cas montrent l’absence de retard de consolidation des fractures des patients sous bisphosphonate.
Ces résultats suggèrent l’absence d’aggravation du statut osseux et permettent l’utilisation des traitements dont l’efficacité sur la fragilité osseuse a été bien démontrée.
La mastocytose
L’ostéoporose est une manifestation classique de la mastocytose qu’il convient de rechercher devant une ostéoporose multifracturaire, en particulier chez un homme ou en présence de manifestations cutanées.
Épidémiologie – La prévalence de la mastocytose est de 3,1 % sur une cohorte de 1 374 patients ostéoporotiques hospitalisés pour fracture. Inversement, dans les cohortes de mastocytose, la prévalence de l’ostéoporose fracturaire varie de 8 à 41 %. La mastocytose vertébrale peut aussi être responsable de lésions condensantes (3 à 8 % des cas) (19).
Point d’appel – Le point d’appel est une tryptase élevée, mais celle-ci est normale chez plus de 80 % des patients avec mutation KIT, comme le souligne le Dr Julien Rossignol.
Manifestations cliniques – Les mastocytoses sont des hémopathies caractérisées par une accumulation de mastocytes atypiques dans un ou plusieurs organes. Les manifestations cliniques de la mastocytose sont liées à l’infiltration des mastocytes et à l’activation des mastocytes. Celle-ci peut se manifester par une anaphylaxie qui est la complication la plus dangereuse, pouvant mettre en jeu le pronostic vital.
Classification – La classification OMS 2022 décrit trois grands types de mastocytose :
• forme cutanée pure, principalement trouvée chez l’enfant ;
• forme systémique, la plus fréquente chez l’adulte
• et le sarcome mastocytaire, une entité rare et de mauvais pronostic.
Parmi les formes systémiques, on distingue :
• les formes avancées de la maladie (qui incluent la forme agressive, leucémique et associée à une autre hémopathie maligne, principalement d’origine myéloïde)
• et les formes non avancées (incluant la mastocytose systémique indolente, médullaire pure et smoldering).
Sur le plan physiopathologique – Les mutations somatiques de gain de fonction dans le gène KIT (en particulier la mutation D816V du gène KIT) sont présentes chez plus de 95 % des patients atteints de mastocytose. Le récepteur KIT (CD117) est un récepteur tyrosine kinase de type III exprimé par les cellules hématopoïétiques, les cellules souches, les cellules germinales, les mélanocytes. Sur les mastocytes, l’activation de ce récepteur, soit lors d’une liaison avec son ligand, soit due à une mutation activatrice, stimule la prolifération, la maturation et la libération des médiateurs pré-formés comme l’histamine ou la tryptase. Récemment, un nouveau trait génétique germinal appelé a-tryptasémie héréditaire (HaT) a été décrit, lié à une duplication/amplification de l’allèle a de TPSAB1 (20). La HaT est associée à un risque plus élevé d’anaphylaxie sévère.
Sur le plan thérapeutique – De nombreux progrès ont eu lieu ces dernières années avec l’utilisation d’inhibiteurs de tyrosine kinase plus ou spécifique du récepteur KIT. Dans les formes non avancées de la maladie, une AMM a été obtenue pour l’avapritinib (inhibiteur de tyrosine kinase ciblant spécifiquement la mutation KIT D816V). Ce médicament a une action sur les symptômes handicapants de la maladie, mais également une action cytoréductrice avec une diminution des lésions cutanées. L’action de l’avapritinib sur l’ostéoporose reste à être étudiée spécifiquement. Dans les formes avancées de la maladie, qui sont caractérisées par un pronostic sombre, la midostaurine (inhibiteur de tyrosine kinase à large spectre) et l’avapritinib ont obtenu une AMM dans cette indication.
Ces traitements ont permis une augmentation de l’espérance de vie des patients, notamment dans les formes agressives. Cependant, une part importante des patients rechute, en particulier avec des formes leucémiques ou associées à une hémopathie maligne, et le seul traitement curatif reste l’allogreffe de cellules souches hématopoïétiques.
Les tumeurs à cellules géantes des gaines téno-synoviales
Les tumeurs à cellules géantes des gaines téno-synoviales (TCG-GTS), anciennement dénommées synovite villo-nodulaire pigmentée, sont une autre classe de tumeur où d’importants progrès thérapeutiques ont été réalisés comme le souligne le Dr Frédéric Paycha.
Physiopathologie – La TCG-GTS est caractérisée par une prolifération synoviale et le développement d’une tumeur composée principalement de cellules géantes multinucléées, de macrophages, d’histiocytes et de cellules inflammatoires. Elle est associée à une expression augmentée du facteur de croissance hématopoïétique CSF-1 (Colony stimulating factor 1). Elle touche l’adulte jeune avec un âge moyen de 30 ans.
Diagnostic et imagerie – Le diagnostic est histologique. L’IRM est l’examen de référence mettant en évidence les dépôts d’hémosidérine au sein de la masse synoviale avec un hyposignal en pondération T1 et T2. La séquence en écho de gradient T2 ou T2* accentue l’artéfact et les dépôts apparaissent alors en signal franc appelé blooming artefact.
Prise en charge – La prise en charge est difficile en raison d’un risque de récidive qui reste important (environ 50 % à 5 ans) malgré l’association chirurgie et synoviorthèse isotopique. La chirurgie exérèse doit être la plus large. Pour le futur, un traitement utilisant le péxidartinib, un inhibiteur tyrosine-kinase de l’axe CSF-1/CSF-1R, pourrait être envisagé. Il a démontré son efficacité dans une étude de phase III, mais était responsable d’effets secondaires graves (21, 22).
La gammapathie monoclonale de signification indéterminée
Le Dr Marina Assadourian fait le point sur la gammapathie monoclonale de signification indéterminée (GMSI), ou mgUS (Monoclonal Gammopathy of Undetermined Significance).
Physiopathologie – La GMSI est secondaire à une prolifération d’un clone lymphoplasmocytaire à l’origine d’une production d’une immunoglobuline monoclonale (IM). La principale crainte est son évolution vers une prolifération maligne lymphoplasmocytaire (myélome multiple (MM), maladie de Waldenström, amylose à chaînes légères, plus rarement des lymphoproliférations de bas grade). Sa prévalence augmente avec l’âge : 3,2 % dans la population âgée de ≥ 50 ans et 5,3 % dans celle âgée de ≥ 70 ans.
Diagnostic – Il s’agit d’un diagnostic d’élimination, retenu devant :
• un pic monoclonal sérique (< 30 g/l) ou urinaire (< 0,5 g/24 h), ou un rapport anormal de chaînes légères libres sériques (CLLs),
• moins de 10 % de plasmocytes dystrophiques clonaux au myélogramme
• l’absence de signes d’atteinte d’organes cibles pouvant être attribuée au trouble prolifératif des plasmocytes clonaux, soit les critères CRAB (hypercalcémie, insuffisance rénale, anémie, lésions ostéolytiques) (23).
Progression – Le risque de progression de la GMSI vers une hémopathie maligne lymphoplasmocytaire est estimé à 1 %. Il dépend de la taille du pic (≥ 15 g/l), du type d’IM (IgM, IgG, IgA, plus rarement IgD, IgE ou à chaînes légères), du rapport des CLLs (k/l), du taux de plasmocytes dystrophiques médullaires et de la présence ou non d’un déficit des autres immunoglobulines. En l’absence de facteur de risque, le risque de progression à 20 ans est de 5 %, mais passe à 21, 37 et 58 % en présence d’un, deux ou trois facteurs de risque, respectivement.
Prise en charge – En pratique, lors de la découverte d’une GMSI, les recommandations actuelles préconisent un contrôle du bilan biologique dans les 3-6 mois, puis le rythme de surveillance dépend du risque de progression : tous les 2 à 3 ans pour les patients avec un faible risque ou tous les 6 mois à 1 an pour les patients avec un risque plus élevé. Le suivi biologique comprend une NFS, une électrophorèse des protéines sériques avec immunofixation, une créatininémie, une calcémie, un dosage des CLLs ainsi qu’une protéinurie totale des 24 h avec immunoélectrophorèse. La réalisation d’un myélogramme ou d’une imagerie osseuse (radiographie, scanner ou IRM corps entier) n’est pas systématique et devra être faite sur point d’appel clinique et/ou facteurs de mauvais pronostic.
Les autres pathologies
Quelles autres actualités ?
La lipomatose épidurale
Une situation clinique dont la prise en charge reste difficile : la lipomatose épidurale.
Épidémiologie – Dr Louis Jacob rappelle que la prévalence de la lipomatose épidurale, due à une accumulation de graisse non encapsulée dans l’espace épidural, est estimée à 2,5 % sur étude américaine de 28 902 patients ayant eu une IRM du rachis. Mais les symptômes étaient concordants dans seulement 5 % des cas. La lipomatose intéressait de façon prépondérante le rachis lombaire (34,5 %) et en moyenne 3,6 niveaux rachidiens.
Manifestations cliniques et diagnostic – Elle peut générer rachialgie, radiculalgie et être responsable de sténose centrale et/ou foraminale. L’IRM est l’examen de référence pour le diagnostic positif et le retentissement neurologique.
Facteurs associés – Il s’agit de l’obésité, de la corticothérapie ou de la production endogène accrue de corticoïdes et d’un antécédent de chirurgie du rachis.
Prise en charge – Elle est difficile, non standardisée. Le traitement médical est à privilégier. Mais les infiltrations sont à discuter et à réserver pour les douleurs persistantes en raison d’une possible aggravation de la lipomatose. La réduction pondérale est recommandée chez des patients obèses. La libération chirurgicale est indiquée en cas de déficit neurologique ou douleur résistante au traitement médical.
Les manifestations rhumatologiques des Covid-19 longs
Une autre situation encore plus difficile, conséquence directe de la Covid-19, les manifestations rhumatologiques des Covid-19 longs. Défi relevé par le Dr Camille Blandin.
Épidémiologie – Défini comme un ensemble de symptômes qui persistent au-delà de 3 mois après une infection à SARS CoV-2, la prévalence du PASC (post-Acute sequelae of SARS CoV-2 infection) en France est estimée à 8 % des patients infectés, ce qui correspond à environ 2 millions de personnes. Elle est plus importante chez des patients qui ont été hospitalisés avec une prédominance féminine.
Diagnostic et présentation clinique – Il s’agit d’un diagnostic d’élimination. Le symptôme principal est l’asthénie persistante après l’infection aiguë, présente dans 40-70 % des cas, associée à des myalgies (13,3 %), des troubles mnésiques (14 %), une dyspnée (10 %), un trouble du sommeil (13 %) et une douleur articulaire (10 %).
Facteurs de risque – Les facteurs de risque de développer un PASC sont le sexe féminin, un asthme préexistant, l’obésité, l’âge, un faible niveau socio-économique, une multi-infection à SARS CoV-2.
La vaccination – L’effet de la vaccination sur l’incidence du Covid long varie selon les études dont certaines montrent une protection et d’autres une absence d’effet.
Prise en charge – La physiopathologie n’est pas connue. Une prise en charge symptomatique est actuellement recommandée, avec réadaptation globale du patient, sans intérêt démontré de la vaccination sur la résolution des symptômes.
Faut-il opérer une coiffe rompue ?
Question traitée par le Pr Johann Beaudreuil, qui nous rappelle qu’il est nécessaire d’avoir à l’esprit :
i) l’existence d’une discordance anatomo-clinique avec, par exemple, amélioration clinique sous traitement conservateur et progression des lésions de la coiffe ;
ii) entre la moitié et les deux tiers des ruptures transfixiantes de la coiffe sont asymptomatiques et une proportion non négligeable de ruptures douloureuses évolue de façon favorable ;
iii) environ 75 % des patients sont améliorés par la massothérapie avec un score de fonction et de qualité de vie maintenu après 5, voire 10 ans de suivi ;
iv) les patients opérés après un échec de la rééducation ont la même qualité de vie que les patients améliorés par la rééducation.
L’ensemble de ces données montre l’importance d’une stratégie de prise en charge graduée médico-chirurgicale graduée, avec, en première intention, un traitement conservateur médical associant traitement antalgique oral, infiltrations cortisoniques, masso-kinésithérapie à base de mobilisations et de sollicitations musculaires et prise en compte des facteurs contextuels (adaptation de l’activité professionnelle, du poste de travail…). Le traitement chirurgical de référence des ruptures de coiffe est la réparation directe. Il est indiqué en l’absence de dégénérescence graisseuse, évaluée sur l’IRM (stade dégénérescence graisseuse ≤ 2, i.e. moins de graisse que de muscle).
Les pathologies du pied
Et, bien sûr, un point sur les pathologies du pied traitées en duo par les Dr Adénike Adedjouma et Virginie Simon.
Le nerf tibial – Plusieurs syndromes canalaires peuvent être observés au pied avec, en particulier, la compression du nerf tibial et de ses branches dans le tunnel tarsien où cheminent également les tendons du muscle tibial postérieur, du long fléchisseur des orteils et du long fléchisseur de l’hallux. Le nerf tibial issu du nerf sciatique à la face postérieure de cuisse devient superficiel au tiers distal de la jambe, le long du bord médial du tendon calcanéen. Il se dirige en arrière de la malléole médiale et s’engage dans le tunnel tarsien où il se divise en deux branches terminales : le nerf plantaire médial et le nerf plantaire latéral. Dans le tunnel, ou juste avant son entrée, le nerf tibial donne naissance au nerf calcanéen qui assure la sensibilité du talon.
Les nerfs plantaires – Le nerf plantaire médial innerve l’abducteur du gros orteil, le court fléchisseur du gros orteil, le court fléchisseur des orteils et le premier muscle lombrical. Le nerf plantaire innerve l’abducteur du 5e orteil, les inter-osseux, l’abducteur du gros orteil et les lombricaux. Les nerfs plantaires assurent la sensibilité plantaire du pied à l’exception du talon.
La compression du nerf tibial – La compression du nerf tibial dans le tunnel tarsien peut être d’origine traumatique, liée à une insuffisance veineuse, une ténosynovite des tendons ou plus rarement une tumeur nerveuse. Elle se révèle par les douleurs neuropathiques et les paresthésies sur les territoires sensitifs associés ou non à un déficit moteur. On peut provoquer un signe de Tinel. La manœuvre d’éversion et de flexion dorsale forcée maintenue pendant quelques secondes permet de déclencher les douleurs et paresthésies. L’électromyogramme et l’échographie permettent de confirmer le diagnostic. Le traitement est médical, corrige les troubles statiques par des orthèses correctrices et éventuellement des infiltrations de dérivés cortisonés. En cas d’échec, une libération chirurgicale peut être discutée.
Autres syndromes canalaires – D’autres syndromes canalaires concernent les compressions des fibulaires superficielle ou profonde, du nerf plantaire médial ou encore du nerf sural.
Allez revoir la communication, les différents dessins anatomiques permettront de mieux les appréhender.
Et l’IA en rhumatologie !
L’intelligence artificielle (IA) a dépassé toutes les prévisions des informaticiens et son utilisation raisonnée permettra d’améliorer nos connaissances et pratiques. Comme le souligne le Pr Thierry Schaeverbeke, il est primordial de s’y investir et de maîtriser ce nouvel outil aux perspectives infinies, même si le taux d’erreurs actuel reste élevé de 40 à 70 % ! Que peuvent raisonnablement en attendre les rhumatologues ?
Chercher une information synthétique sur une maladie, un médicament – Les réponses sont obtenues en quelques secondes, parfaitement structurées. Cette rapidité autorise une utilisation lors d’une consultation, comme on peut interroger le Vidal.
L’aide au diagnostic – ChatGPT est capable, sur un ensemble de signes cliniques et biologiques, de trouver le bon diagnostic dans plus de 70 % des cas en moins de 1 minute. La réponse est argumentée, propose une prise en charge. Plus la description est précise, plus la réponse est fiable.
La traduction d’un texte – Traduction et compréhension d’un texte étaient les premiers objectifs de ChatGPT qui peut maintenant aider la rédaction d’une demande de financement dans n’importe quelle langue… presque !
L’analyse systématique de la littérature – L’IA peut vous aider à faire une méta-analyse ! Elle permet non seulement de retrouver des articles, mais également de capturer des données au sein du texte pour automatiser une partie de l’analyse.
L’aide à la programmation – Chat-GPT est devenu une aide indispensable à la programmation, pour les statistiques complexes ou… pour l’IA !
Ce que peuvent attendre les rhumatologues de l’IA à l’avenir : les premiers succès de l’IA en médecine concernent les analyses d’images (radiologie, anatomopathologie) et en électrophysiologie. Dans le futur, l’IA, en croisant toutes les données de santé disponibles (clinique, biologie, imagerie et traitement) et les données de cohortes prospectives, permettra de prédire l’évolution et le pronostic des maladies, de personnaliser le traitement, de caractériser des groupes de patients à risque, de développer une prévention personnalisée.
Enfin, l’IA sera intégrée à la recherche médicale dans l’optique d’aider à l’identification de nouvelles cibles thérapeutiques, et au développement de nouveaux médicaments.
Qu’on le souhaite ou non, l’IA va évidemment s’imposer dans tous les domaines. Elle fera partie, comme le téléphone portable, des outils que chacun de nous utilisera au quotidien.
L’auteur déclare ne pas avoir de liens d’intérêt en rapport avec cet article.
Bibliographie
1. Müller F, Taubmann J, Bucci L et al. CD19 CAR T-cell therapy in autoimmune disease – a case series with follow-up. N Engl J Med 2024 ; 390 : 687-700.
2. Netea MG, Domínguez-Andrés J, Barreiro LB et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol 2020 ; 20 : 375-88.
3. Gutierrez M, Ruta S, Clavijo-Cornejo D et al. The emerging role of ultrasound in detecting interstitial lung disease in patients with rheumatoid arthritis. Joint Bone Spine 2022 ; 89 : 105407.
4. Cipolletta E, Tata LJ, Nakafero G et al. Association between gout flare and subsequent cardiovascular events among patients with gout. JAMA 2022 ; 328 : 440-50.
5. Ridker PM, Everett BM, Thuren T et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017 ; 377 : 1119-31.
6. Nidorf SM, Fiolet ATL, Arend A et al. Colchicine in patients with chronic coronary disease. N Engl J Med 2020 ; 383 : 1838-47.
7. Tardif JC, Kouz S, Waters DD et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med 2019 ; 381 : 2497-505.
8. Wei J, Choi HK, Dalbeth N et al. Gout flares and mortality after sodium-glucose cotransporter-2 inhibitor treatment for gout and type 2 diabetes. JAMA Network Open 2023 ; 6 : e2330885.
9. McCormick N, Yokose C, Challener GJ et al. Serum urate and recurrent gout. JAMA 2024 ; 331 : 417-24.
10. Pascart T, Robinet P, Ottaviani S et al. Evaluating the safety and short-term equivalence of colchicine versus prednisone in older patients with acute calcium pyrophosphate crystal arthritis (COLCHICORT): an open-label, multicentre, randomised trial. Lancet Rheumatol 2023 ; 5 : e523-31.
11. Latourte A, Ea HK, Frazier A et al. Tocilizumab in symptomatic calcium pyrophosphate deposition disease: a pilot study. Ann Rheum Dis 2020 ; 79 : 1126-8.
12. Cipolletta E, Moscioni E, Sirotti S et al. Diagnosis of calcium pyrophosphate crystal deposition disease by ultrasonography: how many and which sites should be scanned? Rheumatology 2023 : kead565.
13. Mandl P, D’Agostino MA, Navarro-Compán V et al. 2023 EULAR recommendations on imaging in diagnosis and management of crystal-induced arthropathies in clinical practice. Ann Rheum Dis 2024 : ard-2023-224771.
14. Gerwin N, Scotti C, Halleux C et al. Angiopoietin-like 3-derivative LNA043 for cartilage regeneration in osteoarthritis: a randomized phase 1 trial. Nat Med 2022 ; 28 : 2633-45.
15. Epelboym Y, Mandell JC, Collins JE et al. Genicular artery embolization as a treatment for osteoarthritis related knee pain: a systematic review and meta-analysis. Cardiovasc Intervent Radiol 2023 ; 46 : 760-9.
16. Reid IR, Horne AM, Mihov B et al. Fracture prevention with zoledronate in older women with osteopenia. N Engl J Med 2018 ; 379 : 2407-16.
17. Reid IR, Horne AM, Mihov B et al. Effects of zoledronate on cancer, cardiac events, and mortality in osteopenic older women. J Bone Miner Res 2020 ; 35 : 20-7.
18. Jain M, Tam A, Shapiro JR et al. Growth characteristics in individuals with osteogenesis imperfecta in North America: results from a multicenter study. Genet Med 2019 ; 21 : 275-83.
19. Orsolini G, Viapiana O, Rossini M et al. Bone disease in mastocytosis. Immunol Allergy Clin North Am 2018 ; 38 : 443-54.
20. Lyons JJ, Yu X, Hughes JD et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat Genet 2016 ; 48 : 1564-9.
21. Tap WD, Gelderblom H, Palmerini E et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet 2019 ; 394 : 478-87.
22. Lin F, Jacqueline Kwong W, Pan I et al. Real-world patient experience of pexidartinib for tenosynovial giant-cell tumor. Oncologist 2024 ; 29 : e535-43.
23. Kyle RA, Larson DR, Therneau TM et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med 2018 ; 378 : 241-9.