PPR : Peut Parfaitement Répondre – Quels DMARDs, et surtout pour quels patients ?
Baptiste Chevet
Depuis une dizaine d’années, plusieurs essais contrôlés ont démontré l’efficacité de traitements ciblés dans la pseudopolyarthrite rhizomélique (PPR) (1). L’intérêt de ces traitements réside en leur effet cortico-épargneur, c’est-à-dire leur capacité à limiter l’exposition aux corticoïdes et aux effets indésirables inhérents (2). Ces traitements ont toute leur utilité en cas de PPR cortico-dépendante, c’est-à-dire en cas de rechutes répétées lors de la décroissance cortisonée.
Le méthotrexate
Trois essais randomisés ont jusque-là testé le méthotrexate (MTX) à faible dose (7,5-10 mg/sem) dans le cadre de PPR naïve de tout traitement.
L’étude
Au cours de cette édition 2025 du congrès de l’Eular, une étude s’intéressait à l’efficacité du MTX à la posologie de 25 mg/sem par voie orale dans la PPR récemment diagnostiquée (3). Dans les deux bras de traitement, les corticoïdes étaient en décroissance sur 24 semaines. Le critère principal était la rémission (définie par le score d’activité (4) PMR-AS < 10) sans corticothérapie après 1 an de traitement.
Résultats
Les patients du groupe MTX ont été plus nombreux à atteindre l’objectif principal que ceux recevant un placebo (80 versus 46 % ; p = 0,0042), avec un effet toutefois lent, puisqu’il n’y avait pas de différence significative à 32 semaines (46 versus 33 % ; p = 0,21). Le MTX n’a pas montré d’effet cortico-épargneur : il n’y avait pas de réduction significative du nombre de patients rechuteurs dans le bras MTX (60 versus 81 % ; p = 0,10) et il n’a pas été mis en évidence de différence dans la dose cumulée de corticoïdes reçue (2 050 versus 2 288 mg ; p = 0,17).
En pratique
Bien que l’objectif principal ait été atteint, l’absence d’effet cortico-épargneur ne permet pas de se prononcer sur la place du MTX dans l’arsenal thérapeutique. Notons qu’au dernier congrès de l’ACR, en novembre 2024, cette même étude a déjà été présentée, avec des résultats nettement différents : il n’y avait aucune différence dans l’obtention de la rémission entre le groupe MTX et le groupe placebo. L’équipe de travail, interrogée sur ces différences, a expliqué que des erreurs de classification et statistiques lors de la première présentation expliquent la différence forte avec leurs résultats actuels. Les résultats de l’extension de cet essai randomisé sont attendus afin d’évaluer si la rémission est maintenue après 1 an.
Le sécukinumab
Le sécukinumab (SECU) a été évalué chez les patients atteints d’artérite à cellules géantes (ACG) avec des symptômes de PPR, dans une analyse post-hoc de l’étude TitAIN (5).
L’étude TitAIN
Dans cette étude de phase II, les patients étaient randomisés 1:1 entre deux groupes de traitement. Ils bénéficiaient soit de SECU (anti-IL-17A) ou d’un placebo, avec une corticothérapie en décroissance sur 26 semaines. Les patients avec ACG recevant du SECU atteignaient plus la rémission que les patients du groupe placebo. Dans cette analyse post-hoc (6), seuls les patients avec des symptômes de PPR étaient inclus. La PPR était diagnostiquée et suivie cliniquement, sans imagerie dédiée. À l’inclusion dans TitAIN, 12 (44 %) des 27 patients du groupe SECU et 8/25 (32 %) du groupe placebo présentaient des symptômes de PPR.
Résultats
Après 52 semaines, plus de patients recevant du SECU que les patients sous placebo étaient en rémission des symptômes de PPR (58,3 versus 12,5 %), bien que le faible nombre de patients inclus ne permettent pas de conclure à la significativité statistique du résultat.
En pratique
Ainsi, malgré le nombre limité de patients, cette analyse post-hoc montre une réduction des symptômes de PPR jusqu’à huit fois plus fréquente chez les patients traités par SECU. Les données de l’essai REPLENISH, étude de phase III randomisée contre placebo spécifiquement dédiée à la PPR devraient être présentées prochainement.

Bibliographie
1. Kawka L, Chevet B, Arnaud L et al. The pipeline of immunomodulatory therapies in polymyalgia rheumatica and giant cell arteritis: A systematic review of clinical trials. Autoimmun Rev 2024 ; 23 : 103590.
2. Buttgereit F, Matteson EL, Dejaco C, Dasgupta B. Prevention of glucocorticoid morbidity in giant cell arteritis. Rheumatology 2018 ; 57 : ii11-21.
3. Bolhuis T, Kooijman N, Marsman D et al. Randomized double-blind placebo-controlled trial of methotrexate 25mg weekly in newly diagnosed polymyalgia rheumatica. Eular 2025 ; OP0064.
4. Leeb BF, Bird HA. A disease activity score for polymyalgia rheumatica. Ann Rheum Dis 2004 ; 63 : 1279-83.
5. Venhoff N, Schmidt WA, Bergner R et al. Safety and efficacy of secukinumab in patients with giant cell arteritis (TitAIN): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Rheumatol 2023 ; 5 : e341-50.
6. Venhoff N, Schmidt WA, Bergner R et al. Secukinumab in patients with giant cell arteritis with polymyalgia rheumatica symptoms: a post hoc analysis of the phase 2 TitAIN study. Eular 2025 ; OP0062.
7. Wendling D, Al Tabaa O, Chevet B et al. Recommendations of the French Society of Rheumatology for the management in current practice of patients with polymyalgia rheumatica. Joint Bone Spine 2024 ; 91 : 105730.
8. Dejaco C, Singh YP, Perel P et al. 2015 Recommendations for the management of polymyalgia rheumatica: a European League Against Rheumatism/American College of Rheumatology collaborative initiative. Ann Rheum Dis 2015 ; 74 : 1799-807.
9. Chevet B, Drégoire-Perron R, Fromentoux M, Saraux A. Indications of glucocorticoid (GC)-sparing agents in polymyalgia rheumatica: tresholds for cumulative GC dosages based on body weight, occurence of relapse or cortico dependance. Eular 2025 : POS0139.
10. clinicaltrials.gov. University Hospital, Brest. Safety and efficacy of tocilizumab versus placebo in polymyalgia rheumatica with glucocorticoid dependence SEMAPHORE. Disponible sur clinicaltrials.gov/ct2/show/results/NCT02908217 (2020).
11. Dasgupta B, Unizony S, Warrington KJ. Sarilumab in patients with relapsing polymyalgia rheumatica: a phase 3, multicenter, randomized, double blind, placebo controlled trial (SAPHYR). ACR 2022 ; 1676.
Les aspects osseux “difficult-to-manage” au cours des maladies inflammatoires chroniques
Cécile Philippoteaux
Ce paragraphe rapporte la session “difficult-to-manage osteoporosis” du congrès Eular 2025. Inspirée de présentations cliniques de Edgar Wiebe et Frank Buttgereit, elle souligne l’émergence du concept de “difficult-to-treat osteoporosis”, une transposition de la terminologie établie dans les maladies inflammatoires chroniques (polyarthrite rhumatoïde (PR), lupus, etc.).
Ostéoporose et RIC : une étiologie multifactorielle
L’ostéoporose (OP) représente une comorbidité fréquente dans le cadre des maladies rhumatismales inflammatoires. Son origine est multifactorielle, mêlant facteurs “traditionnels” (âge, ménopause, tabagisme…), inflammation systémique chronique, traitements immunomodulateurs (glucocorticoïdes (GC), méthotrexate (MTX)), et altérations métaboliques spécifiques aux maladies de système (malabsorption, vascularite) (Fig. 1) (1, 2).
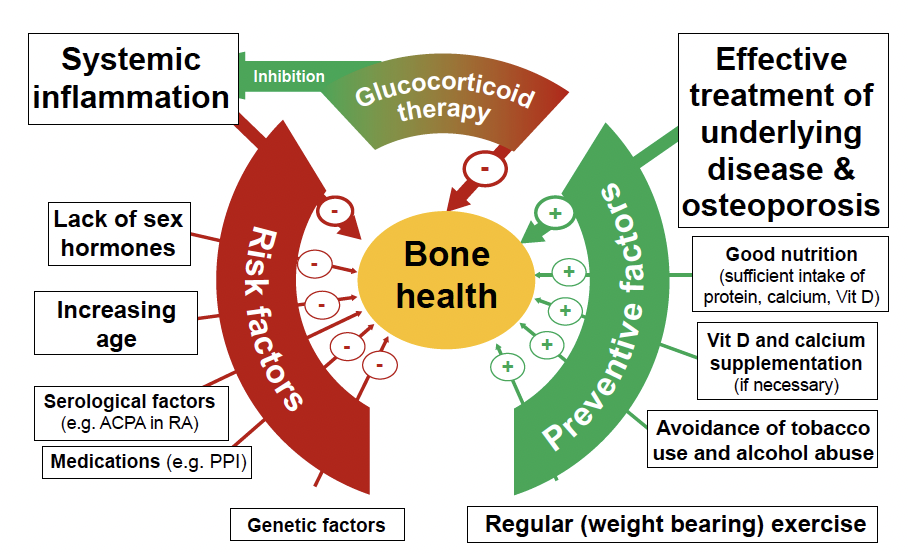
Figure 1 – Aspect multifactoriel de la santé osseuse dans les rhumatismes inflammatoires chroniques (d’après une illustration de Buttgereit (1, 2)).
Ostéoporose dans la sclérodermie systémique
Cas clinique introductif
Une patiente de 40 ans atteinte de sclérodermie systémique (SSc) avec anti-topoisomérase I positive a développé, après plusieurs années de stabilité sous immunosuppresseurs, des calcinoses sévères et des fractures vertébrales spontanées multiples. Malgré une ostéopénie à la densitométrie osseuse biphotonique (DEXA), un scanner quantitatif (QCT) a confirmé une OP sévère. L’introduction d’un traitement ostéo-anabolisant (romosozumab) a permis une stabilisation osseuse, sans aggravation des calcinoses.
Données épidémiologiques et mécanismes
Dans une large cohorte de 485 pa-tients atteints de SSc, Midol et al. ont identifié plusieurs facteurs indépendamment associés à l’OP. Trois facteurs ressortaient de manière significative :
• l’âge (OR = 1,06 ; IC 95 % = 1,04-1,08 ; p < 0,001),
• la positivité des anticorps anti-topoisomérase (OR = 2,22 ; IC 95 % = 1,18-4,16 ; p = 0,013)
• et l’usage de glucocorticoïdes (OR = 4,48 ; IC 95 % = 2,42-8,26 ; p < 0,001) (3).
L’effet des GC dans la SSc est particulier. En effet, à l’inverse de la PR, une faible dose de GC (< 5 mg/j) n’offre pas d’effet protecteur via la réduction inflammatoire, car l’inflammation est modérée ou absente au cours de la SSc (1).
L’usage fréquent d’IPP pour le reflux gastro-œsophagien majore la perte osseuse lorsqu’ils sont combinés aux GC. Une étude transversale menée dans le registre Rh-GIOP (1 495 patients atteints de maladies inflammatoires) a montré que l’usage des IPP est associé à une perte significative de densité minérale osseuse (DMO), mais uniquement en présence concomitante de GC (4).
Enfin, les calcinoses, présentes chez 18 à 25 % des patients SSc, pourraient aggraver la fragilité osseuse (5). Bien que non significative dans l’étude de Midol et al. (3), une cohorte internationale de 5 218 patients a montré une association indépendante entre calcinoses et OP (OR = 4,2 ; IC 95 % = 2,3-7,9 ; p < 0,0001) (6).
Ostéopathie induite par le méthotrexate
Cas clinique introductif
Une femme de 52 ans suivie pour PR, bien contrôlée sous MTX (20 mg/sem) et prednisone (3 mg/j), a présenté des fractures itératives d’insuffisance au niveau des membres inférieurs, en l’absence d’aggravation significative de la DMO à la DEXA. L’IRM a permis d’objectiver des fractures linéaires typiques en bandes, au niveau du tibia distal et du calcanéus, et de retenir le diagnostic d’ostéopathie induite par le MTX. L’arrêt du MTX et l’initiation d’un traitement ostéo-anabolisant ont permis une amélioration clinique.
Caractéristiques cliniques et données récentes
L’ostéopathie induite par le MTX est une entité distincte de l’OP post-ménopausique ou cortisonique. Elle se manifeste par des fractures d’insuffisance atraumatiques, localisées préférentiellement aux os porteurs (tibia, calcanéus) sous forme de fractures linéaires en bandes. Elle touche essentiellement des femmes de 60 ans, avec atteintes bilatérales et signes IRM caractéristiques (7-9).
L’analyse de 30 cas publiés confirme que 100 % des patients présentent des fractures des membres inférieurs, en particulier du tibia distal (53 %) et du calcanéus (13 %) (7-9). Une étude rétrospective récente a évalué 33 patients avec une ostéopathie au MTX (durée moyenne de traitement par MTX de 11 ans, dose moyenne 20 mg/sem) (8). Parmi les 66 % ayant poursuivi le MTX après une fracture initiale, 95 % ont présenté une récidive fracturaire ; chez ceux ayant arrêté le MTX, seuls 36 % ont récidivé (p = 0,001). L’arrêt était associé à une amélioration de la douleur (78 versus 36 %) et de la capacité de mise en charge (71 versus 23 %).
Implications thérapeutiques
La prise en charge repose sur l’arrêt du MTX dès suspicion clinique ou radiologique d’ostéopathie induite au MTX. Les agents ostéo-anabolisants (tériparatide, romosozumab) semblent plus efficaces que les anti-résorptifs (9). Le choix du traitement de fond alternatif doit être individualisé. Des essais prospectifs sont nécessaires pour guider la stratégie thérapeutique.
En pratique
• L’OP et l’atteinte osseuse dans les maladies inflammatoires chroniques peuvent être qualifiées de “difficult-to-treat”.
• Dans la SSc, l’OP est complexe, souvent silencieuse, et est fortement liée à la sévérité de la maladie et ses principaux facteurs de risque incluent l’âge avancé, la positivité des anticorps anti-topo-isomérase I et l’usage de GC.
• L’ostéopathie induite par le MTX doit être reconnue comme une entité distincte, dont la prise en charge repose d’abord sur l’arrêt du MTX, avec recours possible aux traitements ostéo-anabolisants.
Bibliographie
1. Buttgereit F, Palmowski A, Bond M, et al. Osteoporosis and fracture risk are multifactorial in patients with inflammatory rheumatic diseases. Nat Rev Rheumatol 2024 ; 20 : 417-31.
2. Wiebe E, Huscher D, Schaumburg D et al. Optimising both disease control and glucocorticoid dosing is essential for bone protection in patients with rheumatic disease. Ann Rheum Dis 2022 ; 81 : 1313-22.
3. Midol C, Wiebe E, Siegert E et al. Osteoporosis is associated with anti-topoisomerase I positivity and glucocorticoids use in patients with systemic sclerosis. Rheumatology 2025 ; 64 : 1270-6.
4. Palmowski A, Schmajuk G, Yazdany J et al. Proton pump inhibitor use and bone health in patients with rheumatic diseases: a cross-sectional study. Mayo Clin Proc 2024 ; 99 : 1046-57.
5. Davuluri S, Lood C, Chung L. Calcinosis in systemic sclerosis. Curr Opin Rheumatol 2024 ; 36 : 360-9.
6. Valenzuela A, Baron M, Canadian Scleroderma Research Group et al. Calcinosis is associated with digital ulcers and osteoporosis in patients with systemic sclerosis: A Scleroderma Clinical Trials Consortium study. Semin Arthritis Rheum 2016 ; 46 : 344-9.
7. Robin F, Cadiou S, Albert J-D et al. Methotrexate osteopathy: five cases and systematic literature review. Osteoporos Int 2021 ; 32 : 225-32.
8. Hauser B, Merriman A, Foley J et al. Methotrexate continuation increases fracture risk in patients who sustained lower limb insufficiency fractures. Ann Rheum Dis 2025 ; 84 : 554-61.
9. Rolvien T. Methotrexate osteopathy: an increasingly recognised condition manageable only through methotrexate discontinuation. Ann Rheum Dis 2025 ; 84 : 519-20.
Poumon et connectivites : vers un nouveau souffle avec les recommandations Eular/ERS 2025 ?
Margaux Moret
Les atteintes pulmonaires, et en particulier les pneumopathies interstitielles diffuses (PID), sont fréquentes et souvent silencieuses en cas de connectivite, avec un pronostic parfois sévère. Les nouvelles recommandations Eular/ERS 2025 offrent une stratégie pragmatique intégrant le dépistage, le suivi et le traitement dans notre pratique.
Dépister tôt et de façon raisonnée
Le dépistage ne doit pas attendre l’apparition de symptômes, car cela correspond déjà à une démarche diagnostique. Les recommandations insistent sur une approche graduée, adaptée au profil de risque. La TDM thoracique haute résolution (TDM-HR) reste l’examen de référence et ne doit pas être remplacée par les EFR ni par l’échographie pulmonaire, encore en cours d’évaluation.
• Un dépistage systématique par TDM-HR est recommandé chez les patients atteints de sclérodermie systémique (SSc), de connectivite mixte ou de myosite, en particulier en présence de facteurs de risque : syndrome des anti-synthétase, mains de mécanicien, absence d’atteinte musculaire, arthrite, anticorps anti-MDA5 ou anti-Ro52.
• Chez les patients atteints de polyarthrite rhumatoïde (PR) ou de syndrome de Sjögren, le dépistage par scanner est recommandé uniquement si des facteurs de risque sont présents : âge avancé, tabagisme, facteurs rhumatoïdes élevés, présence d’ACPA, syndrome inflammatoire biologique, sexe masculin, activité élevée pour la PR ou âge avancé, syndrome inflammatoire biologique et la présence d’atteintes extra-pulmonaires actives pour le Sjögren.
• Pour chaque patient, il faut rechercher une toux sèche, une dyspnée ou des crépitants à l’auscultation. La réalisation d’EFR avec mesure de la DLCO est indiquée en cas de symptômes ou d’anomalies scannographiques. Toutefois, une DLCO abaissée avec volumes normaux peut évoquer une PID et justifier la réalisation d’un scanner (1).
• En cas de doute diagnostique, un lavage broncho-alvéolaire peut aider à exclure une infection, une hémorragie alvéolaire ou une néoplasie. La biopsie pulmonaire n’est pas retenue pour le diagnostic de PID, mais peut être utile en cas d’atypies à l’imagerie ou pour éliminer une néoplasie.
Suivi des patients : ni trop, ni trop peu, juste ce qu’il faut
Le suivi repose sur une stratification du risque de progression. À chaque étape, il convient d’évaluer les facteurs de mauvais pronostic pulmonaire (Tab. 1).
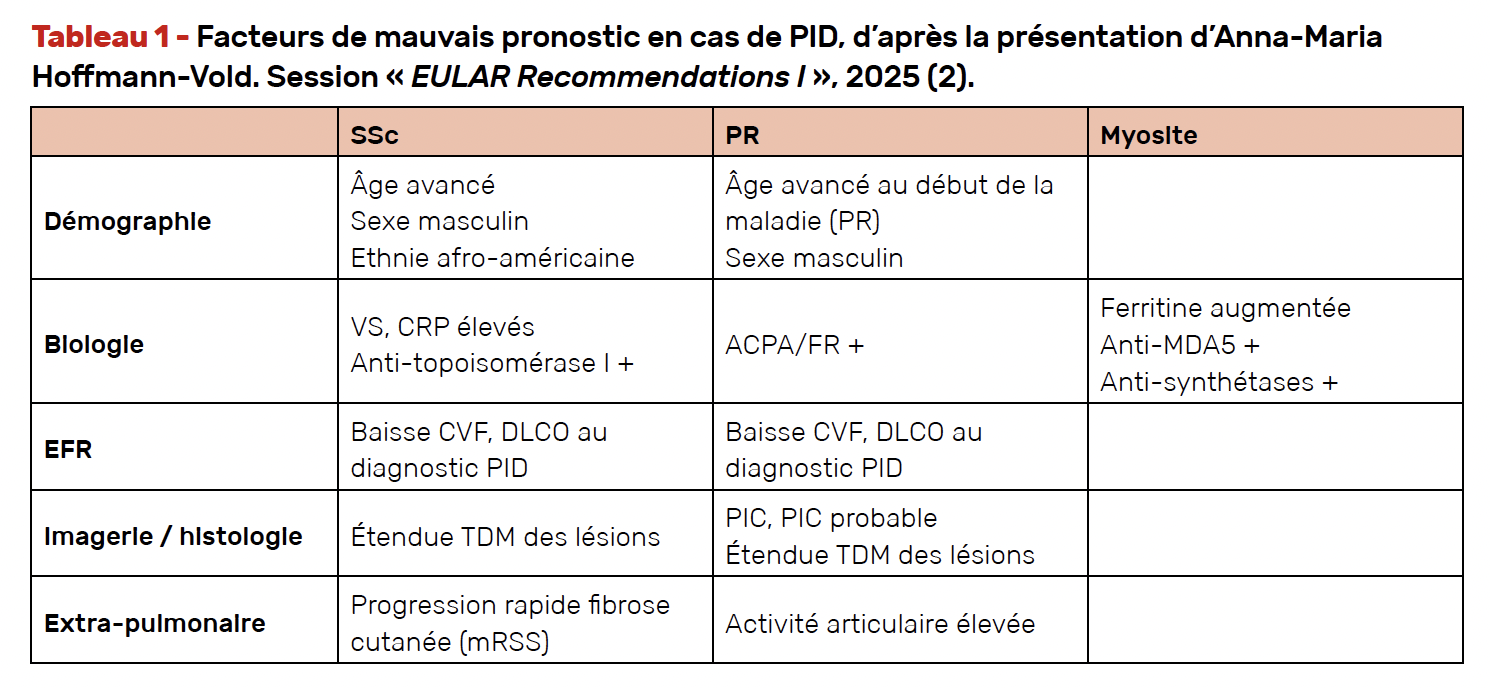
• Les EFR sont à répéter tous les 3 à 6 mois pendant les premières années, puis tous les 6 à 12 mois en cas de stabilité.
• Un scanner de contrôle est recommandé entre 12 et 24 mois, ou plus tôt en cas de signes évolutifs.
• Pour les myosites à haut risque de progression, un contrôle rapproché à 3-6 mois est suggéré.
• Tout signe de progression clinique, fonctionnelle ou radiologique justifie une réévaluation sans délai.
Adapter le traitement au profil évolutif
Sclérodermie systémique (SSc-ILD)
En l’absence de signes cliniques ou radiologiques préoccupants, une surveillance attentive peut suffire.
• En cas d’atteinte pulmonaire étendue ou évolutive, le traitement dépend du niveau d’inflammation, de la fibrose et de l’atteinte systémique.
• En cas de sclérodermie cutanée diffuse précoce avec signes inflammatoires, le tocilizumab ou le mycophénolate mofétil sont recommandés en première intention. L’étude SCLEROLUNG a confirmé l’efficacité du tocilizumab sur la CVF chez les patients avec CRP élevée (3).
• Le nintédanib est indiqué en cas de fibrose étendue > 10 % sur la TDM-HR, sans atteinte extra-pulmonaire.
• En cas de haut risque de progression, une association nintédanib et mycophénolate, ou une combinaison d’immunosuppresseurs peuvent être proposées.
• Pour les formes les plus sévères avec atteinte multiviscérale, des thérapeutiques plus agressives, telles que le cyclophosphamide ou le rituximab, sont recommandées.
Pour rappel, la corticothérapie forte dose est déconseillée.
Polyarthrite rhumatoïde (PR-PID)
• En cas d’activité articulaire persistante, le traitement vise à contrôler l’inflammation systémique, avec une préférence pour abatacept, rituximab ou inhibiteurs de JAK. Le mycophénolate, le tocilizumab ou l’azathioprine peuvent également être utilisés.
• Si la PR est contrôlée, mais que la PID progresse, en particulier sur le plan fibrosant ou en cas de PIC, une intensification est justifiée : combinaison d’immunosuppresseurs, ajout d’un antifibrosant comme le nintédanib, voire pirfénidone (4).
La corticothérapie à forte dose au long cours est à éviter.
Myosites
La corticothérapie, les inhibiteurs de calcineurine et le rituximab sont les piliers du traitement, le mycophénolate ou l’azathioprine peuvent également être envisagés.
• En cas de haut risque de progression et/ou d’atteinte multiviscérale, ces traitements peuvent être associés en ajoutant également les IgIV à l’arsenal thérapeutique.
• Dans les formes rapidement progressives, les inhibiteurs de JAK ou la plasmaphérèse peuvent être considérés.
• Le nintédanib est recommandé dans les PID fibrosantes progressives, seul ou associé à une immunosuppression.
Des recommandations ont également été établies pour les autres connectivites, mais ne sont pas présentées ici. Pour tous, en cas de progression ou de réponse insuffisante, une réévaluation thérapeutique est nécessaire et la discussion multidisciplinaire avec les pneumologues reste essentielle. Dans les formes les plus graves, l’orientation vers une évaluation pré-transplantation pulmonaire est parfois nécessaire.
En pratique
En somme, même si certaines recommandations restent conditionnelles et nécessitent d’être consolidées par de nouvelles études, l’Eular encourage une approche structurée : un dépistage précoce et raisonné, un traitement adapté au risque évolutif, et un suivi rigoureux.
Bibliographie
1. Hoffmann AM, Rodziewicz M, Mehta P et al. Screening and monitoring of ILD in connective tissue diseases – EULAR recommendations. Ann Rheum Dis 2023 ; 2024 ; 8 : rkae056.
2. EULAR/ERS diagnostic and therapeutic approach of interstitial lung disease in connective tissue diseases. Ann Rheum Dis 2025 ; Epud ahead of print.
3. Distler O et al. Update on treatment algorithms in SSc-ILD. Eular 2025 : LB001.
4. Flaherty KR, Wells AU, Cottin V et al. Nintedanib in progressive fibrosing intersitial lung diseases. N Engl J Med 2019 ; 381 : 1718-27.
Mise au point sur la polychondrite atrophiante
Maëva Masson
Lors du congrès Eular 2025, le Pr Laurent Arnaud (Strasbourg) a proposé une mise au point brillante et actualisée sur la polychondrite atrophiante (PCA), une maladie rare, souvent méconnue, mais dont l’intérêt a été ravivé par la découverte du syndrome VEXAS, auquel elle est fréquemment associée.
Points clés sur la maladie
Sa prévalence est estimée à 9/million d’habitants, avec un âge de début entre 40 et 60 ans et un sex-ratio équilibré (1). La maladie touche principalement le cartilage de l’oreille (90-95 %), du nez (50-70 %), de la trachée (30-50 %) et du sternum (10-30 %), avec, à terme, des risques de déformation estimés à 10 %.
Reconnaître et distinguer la chondrite
La chondrite auriculaire est l’une des présentations les plus typiques de la PCA. Elle se manifeste par un érythème, un œdème douloureux de l’oreille, touchant la partie cartilagineuse et épargnant toujours le lobule. L’épisode dure généralement plus de 48 heures, souvent 1 à 2 semaines. Plusieurs diagnostics peuvent mimer une chondrite, parmi lesquels on peut citer :
• érysipèle ou cellulite, notamment en cas de porte d’entrée comme un piercing ;
• otite externe dans un contexte immunodéprimé, souvent avec otorrhée ;
• maladie de Lyme (lymphocytome borrélien) ;
• lymphome T ;
• syndrome catastrophique des anti-phopholipides (aspect purpurique, nécrotique) ;
• autres : piqûre d’insecte, érythème solaire ou engelure, troubles vasomoteurs.
Autres diagnostics en présence d’une chondrite non liée à la PCA
La PCA n’est pas la seule cause de chondrite. D’autres pathologies inflammatoires ou auto-immunes peuvent également provoquer une atteinte cartilagineuse.
• Granulomatose avec polyangéite (GPA) : souvent ANCA positive, avec atteinte rénale (non classique dans la PCA), ORL, et sténose sous-glottique.
• Syndrome VEXAS : chondrite fréquente, souvent sévère, chez l’homme > 60 ans, avec signes myéloïdes associés.
• De façon plus rare : lupus systémique érythémateux, sarcoïdose, maladie liée aux IgG4.
Atteinte trachéale : gravité et pièges diagnostiques
Elle peut se manifester par une dysphonie, un stridor, une douleur cervicale ou thoracique, voire une détresse respiratoire. Le scanner en inspiration/expiration permet de détecter un épaississement circonférentiel de la trachée, respectant la membrane postérieure, habituellement localisé et sous-glottique, ce qui la distingue de la GPA. Sauf exception, la fibroscopie est contre-indiquée en raison du risque élevé de perforation (2).
Les atteintes extra-cartilagineuses
Les manifestations systémiques précèdent parfois l’atteinte cartilagineuse, rendant le diagnostic initial difficile :
• atteinte articulaire (≈ 75 %) : séronégative, migratoire, asymétrique, axiale possible, non érosive, sans nodules ;
• atteinte oculaire : épisclérite, sclérite, scléromalacie ;
• atteinte cochléo-vestibulaire : hypoacousie, vertiges ;
• atteinte cutanée : nodules aux membres inférieurs, purpura vasculaire ;
• atteinte cardiovasculaire : valvulopathie (3).
Le syndrome VEXAS doit être suspecté en cas de fièvre inexpliquée, myélodysplasie ou signes indirects (macrocytose, thrombopénie), d’atteinte cutanée type dermatose neutrophilique, d’atteinte cardiovasculaire ou d’infiltrat pulmonaire chez un homme > 60 ans (4).
Les examens complémentaires
La biologie est peu spécifique :
• CRP normale dans 30-40 % ;
• anticorps anti-nucléaires (20 %), facteurs rhumatoïdes (17 %), anticorps anti-CCP (4 %) ;
• ANCA négatifs.
La recherche d’anticorps anti-cartilage, anti-collagène de type 2 et/ou anti-matrilline 1 est inutile. La biopsie cartilagineuse est rarement nécessaire. Les examens utiles incluent un scanner cervico-thoracique, EFR, échographie cardiaque, et radiographies articulaires. Le caractère dynamique du scanner permet de dépister une éventuelle trachéo-bronchomalacie (collapsus expiratoire). Des érosions du cartilage laryngé ou trachéal visibles au scanner peuvent constituer des preuves indirectes en faveur du diagnostic. L’analyse d’images rétrospectives (photos) peut être également précieuse.
Prise en charge
La majorité des patients reçoivent des corticoïdes, souvent à hautes doses, mais les rechutes sont fréquentes si aucun traitement de fond n’est associé (5, 6).
Le traitement dépend de la sévérité :
• épisode mineur : AINS ± prednisone ;
• rechutes : colchicine (pendant au moins 6 mois), méthotrexate, dapsone ;
• formes sévères ou systémiques : biothérapies (anti-TNF, anti-IL-6, ± inhibiteurs de JAK) d’emblée.
Malgré les biothérapies, moins de 50 % des patients atteignent une réponse complète sous biothérapie (7). L’atteinte trachéale est particulièrement difficile à contrôler, souvent chronique et récidivante (8). Des mesures de support peuvent être proposées : traitement par laser (recommandé en première intention en cas d’atteinte trachéale), prothèse ou stents trachéaux, implants cochléaires si surdité ou rhinoplastie reconstructrice en cas de séquelles esthétiques.
Bibliographie
1. Hazra N, Dregan A, Charlton J et al. Incidence and mortality of relapsing polychondritis in the UK: a population-based cohort study. Rheumatol 2015 ; 54 : 2181-7.
2. Wang ST, Wang J, Gao X et al. Risk factors associated with severe adverse events in patients with relapsing polychondritis undergoing flexible bronchoscopy. Orphanet J Rare Dis 2024 ; 19 : 54.
3. Shimizu J, Oka H, Yamano Y et al. Cardiac involvement in relapsing polychondritis in Japan. Rheumatol 2016 ; 55 : 583-4.
4. Khitri MY, Guedon AF, Georgin-Lavialle S et al. Comparison between idiopathic and VEXAS-relapsing polychondritis: analysis of a French case series of 95 patients. RMD Open 2022 ; 8 : e002255.
5. Yoshida T, Yoshifuji H, Shirakashi M et al. Risk factors for the recurrence of relapsing polychondritis. Arthritis Res Ther 2022 ; 24 : 127.
6. Sangle SR, Hughes CD, Barry L et al. Relapsing polychondritis – A single Centre study in the United Kingdom. Autoimmun Rev 2023 ; 22 : 103352.
7. Yang R, Rhee RL, Quinn KA et al. Clinical manifestations and treatment in patients with relapsing polychondritis: a multicenter observational cohort study. ACR Open Rheumatol 2025 ; 7 : e70027.
8. Moulis G, Pugnet G, Costedoat-Chalumeau N et al. Efficacy and safety of biologics in relapsing polychondritis: a French national multicentre study. Ann Rheum Dis 2018 ; 77 : 1172-8.
Les auteurs déclarent ne pas avoir de liens d’intérêt.